Clear Sky Science · pt
Modelo de aprendizado analítico orientado por genes para diagnóstico preciso do câncer de mama
Por que esta pesquisa importa para pacientes e famílias
O câncer de mama é hoje o câncer mais comumente diagnosticado em mulheres em todo o mundo, e pacientes que parecem ter a mesma doença no papel podem ter desfechos muito diferentes. Este estudo mostra como padrões em milhares de genes, combinados com um sistema de inteligência artificial cuidadosamente projetado, podem ajudar médicos a distinguir com maior confiabilidade quem tem câncer e quão agressivo ele pode ser—usando apenas dados reais de pacientes e um conjunto compacto de genes-chave.
De muitos fatores de risco à linguagem dos genes
O risco de câncer de mama é moldado por muitas influências: alterações genéticas herdadas, hormônios, peso corporal, estilo de vida e mais. Uma vez que o câncer aparece, seu comportamento é determinado por quais genes estão ativados ou desativados dentro de cada tumor. O sequenciamento moderno pode medir a atividade de dezenas de milhares de genes ao mesmo tempo, mas transformar esse oceano de números em respostas claras de sim ou não para diagnóstico e prognóstico é difícil. Métodos computacionais tradicionais frequentemente analisam genes um a um e podem perder a forma como grupos de genes atuam em conjunto, ou podem ter bom desempenho apenas em um conjunto de dados e falhar quando testados em outro.
Treinando um modelo de dupla "mente" para ler padrões gênicos
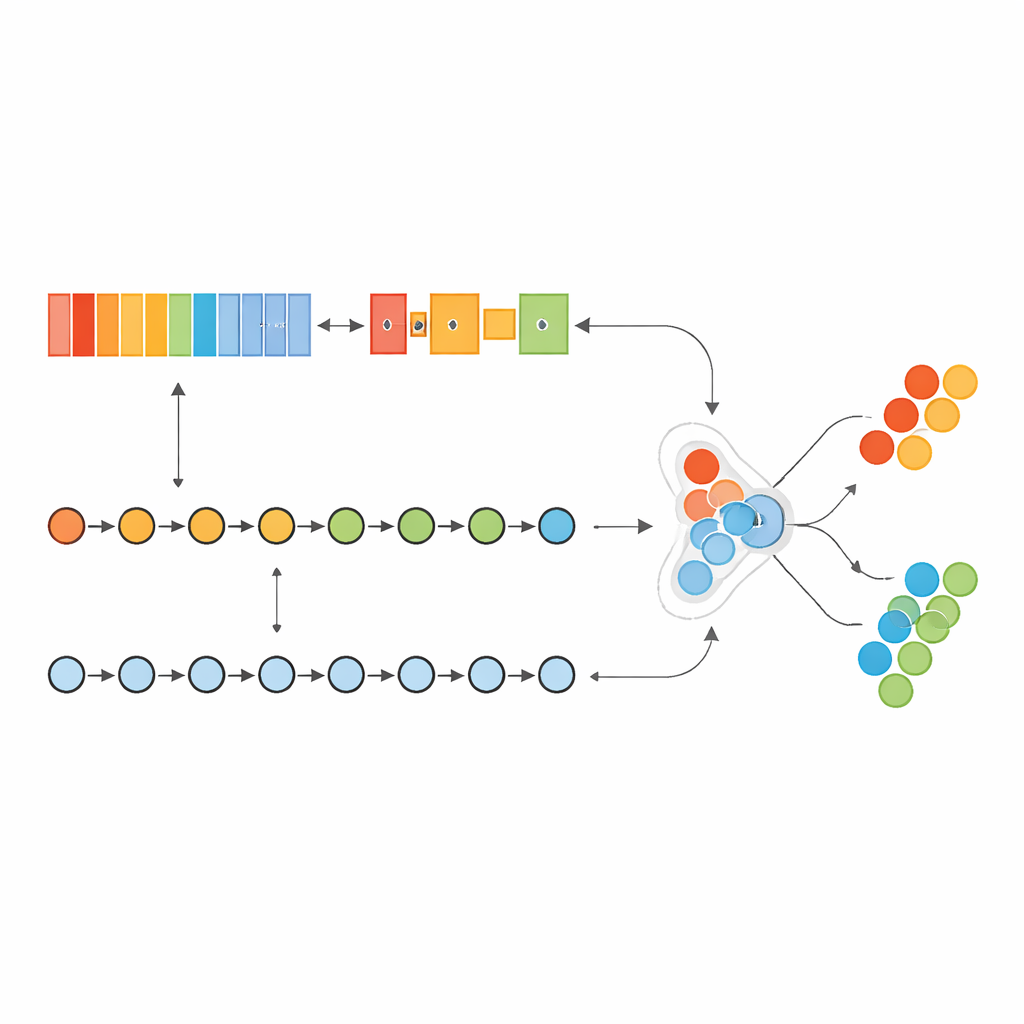
Os autores construíram um modelo híbrido de aprendizado profundo que age um pouco como duas mentes especializadas trabalhando juntas. Uma parte, inspirada na análise de imagens, varre uma lista ordenada de genes para detectar padrões locais—aglomerados de genes cuja atividade em conjunto sinaliza câncer. A outra parte trata os mesmos genes como uma sequência, aprendendo como genes “condutores” iniciais e genes “a jusante” posteriores influenciam uns aos outros ao longo da lista. Ao combinar essas duas visões, o modelo consegue capturar tanto relações de curto alcance quanto de longo alcance dentro da impressão digital genética do tumor.
Encontrando um conjunto central estável de genes sinal
Em vez de alimentar todas as 17.815 genes medidos no modelo, a equipe projetou um pipeline rigoroso e "livre de vazamento" para selecionar apenas os mais informativos. Usando uma medida padrão de correlação dentro de laços de verificação cruzada repetidos, eles classificaram repetidamente os genes pela força com que sua atividade se correlacionava ao estado de câncer. Em seguida, mantiveram apenas os genes que consistentemente surgiam no topo em todas as divisões de treinamento, resultando em uma assinatura estável de 236 genes. Os pesquisadores também mapearam como esses genes interagem entre si, mostrando que muitos formam redes fortemente conectadas relacionadas ao crescimento tumoral, metabolismo, imunidade e ao ambiente tecidual circundante—evidência de que o conjunto escolhido reflete biologia real, não ruído aleatório.
Colocando o modelo à prova
O sistema híbrido foi treinado e ajustado em amostras de câncer de mama do The Cancer Genome Atlas e então desafiado com um conjunto de dados inteiramente separado conhecido como METABRIC. Para lidar com o fato de que amostras de câncer superam em muito as amostras normais, os autores não criaram dados artificiais; em vez disso, ajustaram quanto o modelo "se importa" com erros na classe mais rara. Após uma busca automatizada pelos melhores parâmetros, o modelo alcançou pontuações quase perfeitas em seu conjunto de dados principal, identificando corretamente quase todos os casos de câncer e gerando virtualmente nenhum alarme falso. Importante, o desempenho permaneceu extremamente alto e muito estável mesmo quando o modelo foi aplicado à coorte externa METABRIC, sugerindo que a abordagem pode generalizar além de um único estudo ou hospital.
O que isso significa para o cuidado futuro
Em termos simples, este trabalho entrega uma IA finamente ajustada, em duas partes, que lê um código compacto de 236 genes para distinguir amostras de mama cancerosas de não cancerosas com notável precisão e consistência, mesmo em condições ruidosas. Embora o estudo atual examine apenas a atividade gênica e use dados passados de pacientes, seus métodos preparam o terreno para ferramentas futuras que poderiam combinar múltiplos tipos de dados—como imagens de tecido e camadas moleculares adicionais—e fornecer explicações claras de quais genes impulsionam cada predição. Com validação adicional em estudos clínicos prospectivos, tal sistema poderia se tornar uma espinha dorsal universal para o diagnóstico de precisão do câncer de mama, ajudando médicos a ajustar tratamentos usando a "assinatura" genética do tumor de cada paciente.
Citação: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Palavras-chave: diagnóstico do câncer de mama, expressão gênica, aprendizado profundo, CNN-BiLSTM, oncologia de precisão