Clear Sky Science · pt
CiCLoDS: Agrupamento conjunto de células e seleção de genes para transcriptômica espacial unicelular
Encontrando Vizinhanças na Cidade das Células
Microscópios modernos já conseguem detectar quais genes estão ativos em centenas de milhares de células mantendo cada célula em sua posição original dentro do tecido. Essa revolução da “transcriptômica espacial” é como transformar um mapa urbano borrado em uma vista ao nível da rua de cada casa. Mas há um problema: esses mapas contêm medições de milhares de genes por célula, muito mais do que os cientistas conseguem interpretar facilmente ou custear em experimentos de seguimento. Este estudo apresenta o CiCLoDS, um novo método que encontra vizinhanças celulares significativas e, ao mesmo tempo, seleciona uma lista pequena e interpretável de genes que definem essas vizinhanças.
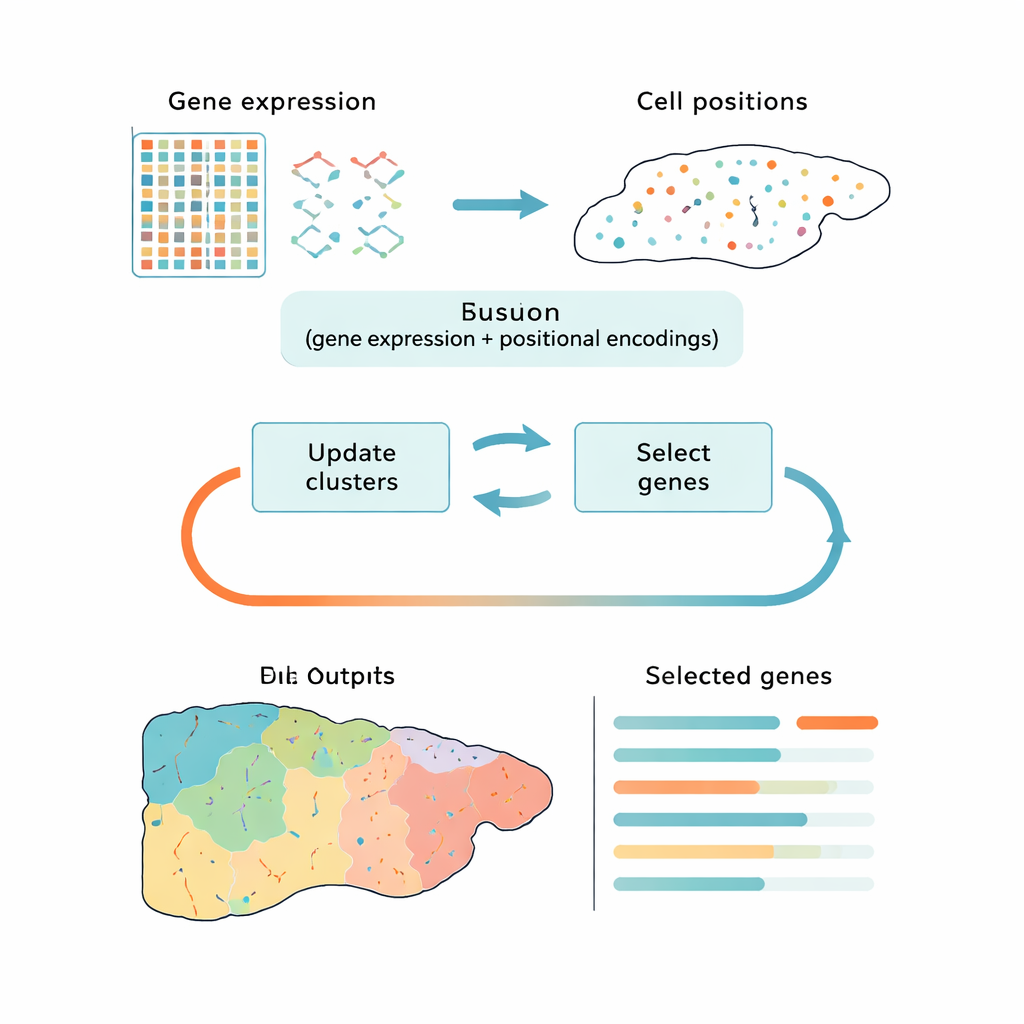
Uma Maneira Mais Inteligente de Reduzir Grandes Dados
A maioria das ferramentas atuais lida com esse desafio em dois passos desconectados: primeiro reduzem os dados a uma forma mais simples e depois agrupam as células em clusters. Abordagens populares como análise de componentes principais (PCA) preservam a variação geral, mas podem enfatizar ruído técnico ou sinais genéricos do ciclo celular em vez das diferenças biológicas relevantes. Outros métodos usam aprendizado profundo para encontrar padrões, mas funcionam como caixas‑pretas e não mostram claramente quais genes são mais importantes. O CiCLoDS segue uma rota diferente. Ele trata seleção de genes e agrupamento como um único problema conjunto sob um “orçamento” definido pelo usuário para quantos genes podem ser mantidos. Em essência, pergunta: qual conjunto limitado de genes explica melhor como as células se organizam em grupos distintos, considerando tanto sua atividade gênica quanto, quando disponível, suas posições físicas no tecido?
Da Matemática aos Mapas de Tecidos Reais
Os autores adaptam uma família de técnicas matematicamente transparentes chamadas subspace clustering às exigências da transcriptômica espacial, onde conjuntos de dados podem conter mais de um milhão de células. O CiCLoDS funciona sobre uma tabela simples célula‑por‑gene, atribuindo células a clusters enquanto pontua cada gene segundo o quanto ele ajuda a separar esses clusters. Ele também pode integrar informação espacial adicionando “encodings” posicionais que descrevem onde cada célula se localiza no tecido, sem alterar a otimização central. Em grandes conjuntos de dados de fígado de camundongo e cólon humano gerados por plataformas de imagem de alta resolução, o CiCLoDS roda em minutos em computadores padrão e produz painéis de genes compactos — na ordem de poucas dezenas a algumas centenas de genes — que ainda capturam a estrutura rica dos dados originais.
Revelando Zonas Ocultas e Vasos Sanguíneos
Ao aplicar o CiCLoDS ao fígado de camundongo, a equipe testou se o método recuperava padrões conhecidos de “zonation” — mudanças graduais na função dos hepatócitos de um lado do lóbulos ao outro. Em comparação com PCA e uma ferramenta líder de seleção de genes chamada geneBasis, o CiCLoDS produziu zonas espaciais mais nítidas, com limites mais definidos e muito menos regiões mal atribuídas, conforme mostram métricas quantitativas que medem concordância com um mapa de referência. Notavelmente, quando autorizado a usar mais genes, o CiCLoDS redescobriu grupos de hepatócitos com características peri‑portal e peri‑central que corresponderam de perto a clusters de referência definidos por especialistas, mesmo sem conhecer o gene biomarcador-chave AXIN2 ou receber coordenadas espaciais explícitas. Quando os encodings espaciais foram adicionados, o CiCLoDS também aprendeu painéis de genes enriquecidos para funções de superfície celular e relacionadas a vasos, e conseguiu distinguir com precisão vasos sanguíneos reais de artefatos de imagem — algo que métodos mais simples ou falhavam em fazer ou só alcançavam com ajustes ad‑hoc maiores.
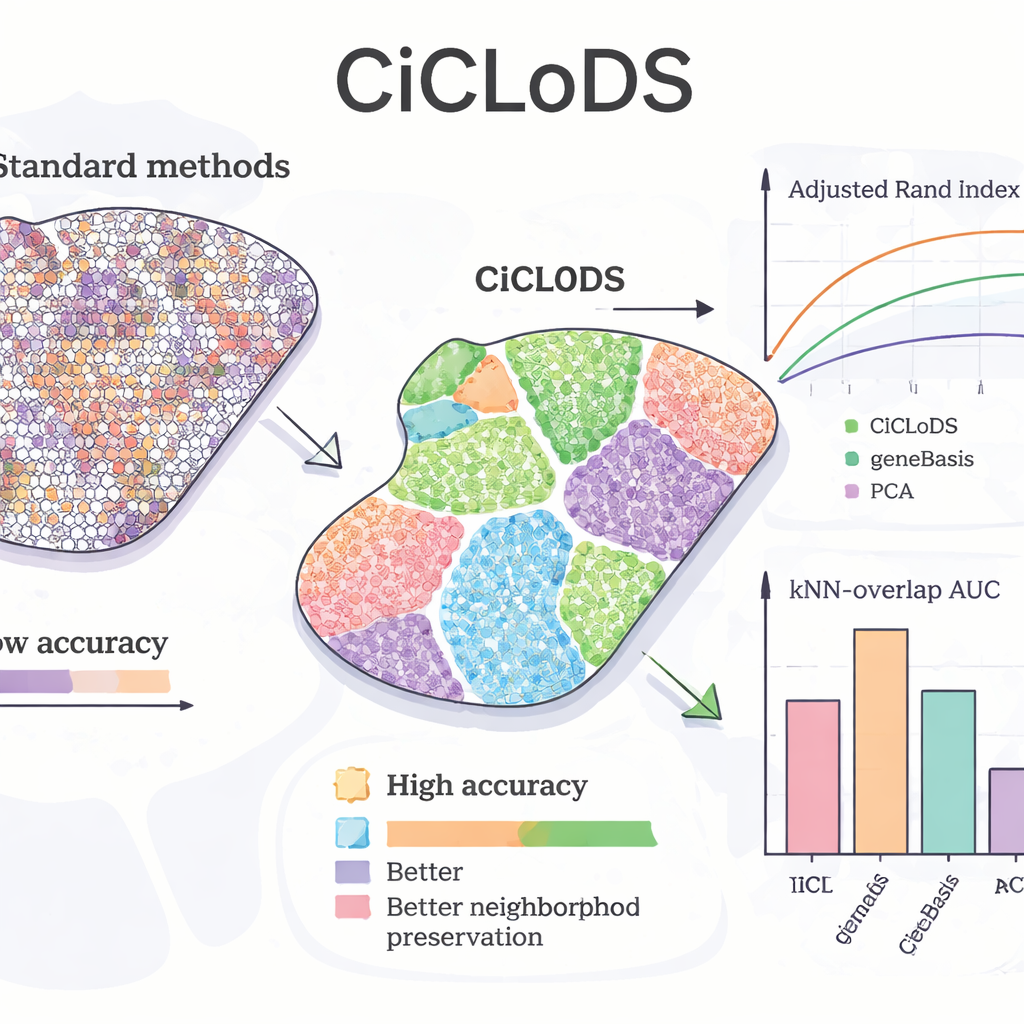
Generalizando Entre Cérebros e Melhorando Outros Métodos
Para avaliar se o CiCLoDS se mantém em tecidos e indivíduos muito diferentes, os autores analisaram amostras do córtex pré‑frontal dorsolateral humano de três doadores. Aqui, o CiCLoDS teve desempenho comparável ou superior a métodos espaciais especializados como BayesCafe e BayesSpace, particularmente em uma amostra difícil na qual as outras ferramentas apresentaram dificuldades. O estudo também destaca um uso “híbrido”: rodar o CiCLoDS primeiro para obter clusters estáveis e depois alimentar esses resultados no BayesSpace. Essa estratégia de warm‑start aumentou a acurácia geral e produziu padrões de camadas cerebrais que melhor corresponderam às anotações de especialistas, mostrando que o CiCLoDS pode tanto atuar de forma autônoma quanto tornar modelos probabilísticos subsequentes mais confiáveis.
Por Que Isso Importa para Biologia e Medicina
Para não‑especialistas, a principal conclusão é que o CiCLoDS transforma mapas celulares avassaladores em resumos concisos e biologicamente relevantes. Em vez de trabalhar com milhares de medições ruidosas, os pesquisadores obtêm uma lista manejável de genes e clusters espaciais claros que refletem a organização real do tecido — zonas metabólicas no fígado, vasos sanguíneos e seus nichos, e estruturas em camadas no cérebro. Como o orçamento de genes é controlado pelo usuário e os cálculos são leves, o CiCLoDS pode ajudar a projetar painéis de genes direcionados para experimentos futuros, orientar a interpretação de conjuntos de dados espaciais complexos e fornecer pontos de partida robustos para modelagens mais elaboradas. Em uma era em que o gargalo não é mais a coleta de dados, mas o entendimento, ferramentas como o CiCLoDS prometem tornar mapas teciduais de alta dimensão ao mesmo tempo práticos e perspicazes.
Citação: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Palavras-chave: transcriptômica espacial, agrupamento de células, seleção de painel de genes, arquitetura tecidual, análise unicelular