Clear Sky Science · pt
Controle orbital da condutividade térmica eletrônica em monocamada de h-B2O via dopagem no regime difusivo
Por que uma folha de um átomo importa para o calor
À medida que nossos celulares, laptops e futuros dispositivos quânticos encolhem, dissipar calor torna‑se um dos maiores desafios de engenharia. Este artigo explora um novo material ultrafino chamado borofeno óxido em favo de mel (h‑B2O), com apenas um átomo de espessura, que conduz calor de maneira incomum e altamente direcional. Ao entender e controlar como os elétrons transportam calor através dessa folha, os cientistas esperam projetar componentes minúsculos que espalhem o calor de forma eficiente ou, deliberadamente, o mantenham confinado para dispositivos de conversão de energia. 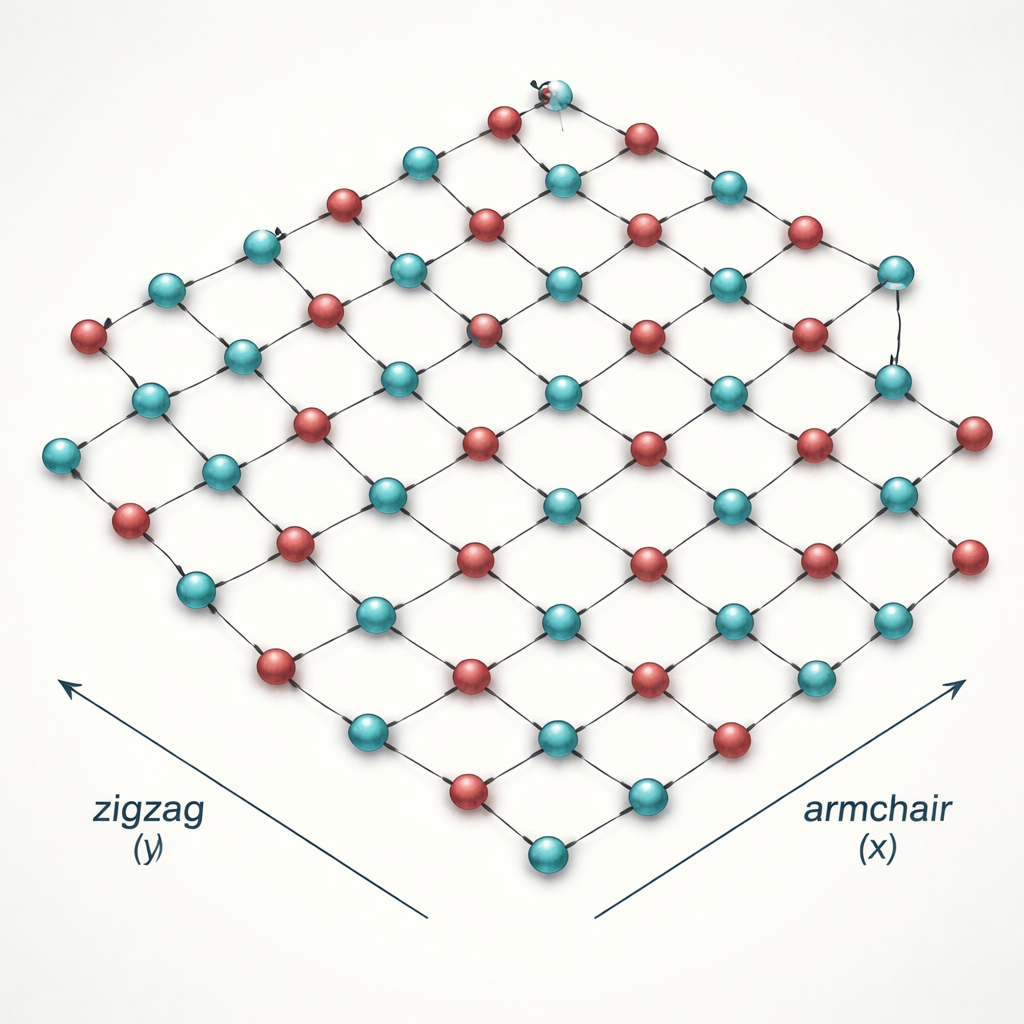
Um novo parente do grafeno
Desde a descoberta do grafeno, pesquisadores buscam outros cristais de um átomo de espessura com propriedades eletrônicas e térmicas especiais. O boro, vizinho do carbono, pode formar suas próprias redes planas chamadas borofeno, e quando átomos de oxigênio são adicionados de maneira adequada, o resultado é h‑B2O, uma folha estável, perfeitamente plana e em favo de mel. Estudos anteriores mostraram que esse material é mecanicamente robusto, pode abrigar estados eletrônicos exóticos chamados laços nodais e pode até se tornar supercondutor a baixas temperaturas. Isso torna o h‑B2O uma plataforma promissora para eletrônica de próxima geração, armazenamento de hidrogênio e catálise, se seu comportamento térmico for plenamente mapeado.
Seguindo os elétrons, não apenas as vibrações
Nos sólidos, o calor pode se propagar de duas maneiras principais: por meio da vibração dos átomos (fônons) e por meio de elétrons em movimento. Para o h‑B2O, a contribuição baseada em vibrações já havia sido calculada, mas a parcela eletrônica permaneceu desconhecida. Os autores constroem um modelo matemático simplificado porém preciso que foca em dois estados eletrônicos específicos dos átomos de boro, chamados orbitais Py e Pz. Esses dois “canais” dominam o comportamento dos elétrons perto dos níveis de energia relevantes para transporte. Usando uma abordagem quântica conhecida como formalismo de Kubo–Greenwood, eles calculam quanto calor os elétrons podem transportar em três direções: ao longo de um eixo da rede (“armchair”), ao longo do outro (“zigzag”) e transversalmente, em um efeito análogo à resposta térmica de Hall. 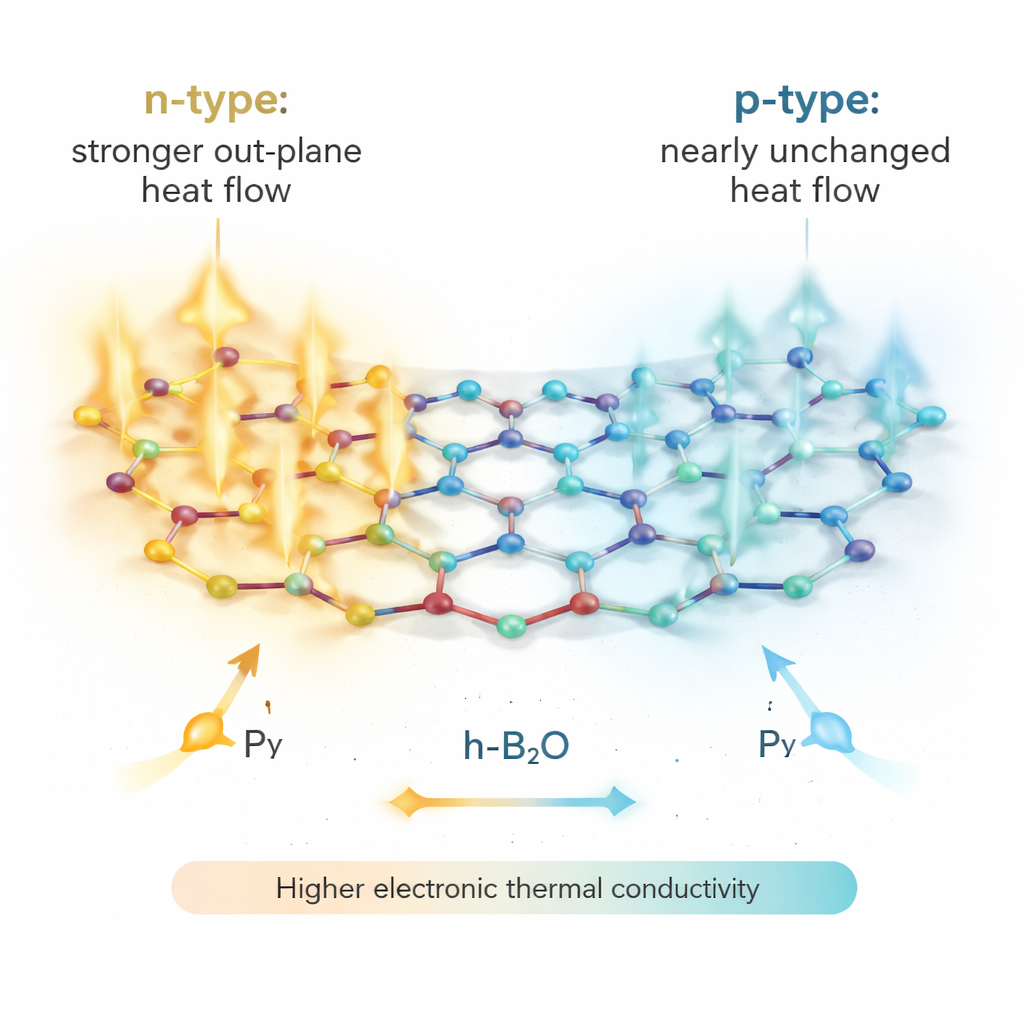
O calor prefere uma direção e um orbital
Os cálculos revelam que o fluxo de calor eletrônico no h‑B2O é fortemente assimétrico: na direção zigzag é muito maior do que na direção armchair. Essa preferência direcional decorre de sutis distorções no padrão hexagonal, que alteram a intensidade das interações entre átomos de boro vizinhos. Elétrons que viajam por caminhos zigzag encontram “rodovias” mais eficientes, enquanto os que seguem armchair enfrentam mais resistência. O orbital Pz, que se projeta fora do plano, fornece mais estados eletrônicos disponíveis perto dos níveis de energia-chave e permite que os elétrons se movam com mais facilidade, portanto carrega a maior parte do calor eletrônico. O orbital Py, no plano, contribui muito menos, embora ainda seja importante para moldar a estrutura eletrônica global.
Virando um botão térmico com impurezas
Dispositivos reais nunca são perfeitamente limpos, então a equipe estuda como impurezas adicionadas — átomos extras ou defeitos que doam elétrons (tipo n) ou os removem (tipo p) — alteram o transporte térmico eletrônico. Usando uma técnica chamada método T‑matrix para tratar a dispersão por essas impurezas, eles constatam que a dopagem tipo n na verdade aumenta a condutividade térmica eletrônica, especialmente através do canal Pz. Adicionar elétrons preenche estados fora do plano que funcionam como faixas extras para elétrons que carregam calor, enquanto o canal Py torna‑se ligeiramente mais localizado e menos efetivo. O fluxo térmico eletrônico total aumenta em todas as direções, embora de forma desigual. Em contraste, a dopagem tipo p produz apenas mudanças modestas: Py ganha um pouco, Pz perde um pouco, e o transporte térmico eletrônico global permanece quase inalterado e estável em uma faixa de temperaturas e níveis de impurezas.
O que isso significa para dispositivos futuros
Para um não‑especialista, a mensagem é que o h‑B2O se comporta como um fio térmico atômico altamente direcional e ajustável. Seus elétrons preferem transportar calor ao longo de uma direção in‑plane e principalmente por um canal orbital específico. Ao escolher como dopar o material — adicionando impurezas doadoras de elétrons ou geradoras de lacunas — os engenheiros podem tanto aumentar fortemente esse fluxo térmico eletrônico (com dopagem tipo n) quanto mantê‑lo quase inalterado (com dopagem tipo p). Combinado com sua estabilidade estrutural e estados eletrônicos incomuns, isso faz da monocamada de h‑B2O um forte candidato para módulos termelétricos em escala nanométrica que convertem calor residual em eletricidade, bem como para elementos de gerenciamento térmico em chips projetados para direcionar calor para longe ou em direção a regiões específicas de um dispositivo.
Citação: Mohammadi, F., Mirabbaszadeh, K. & Noshad, H. Orbital-resolved tuning of electronic thermal conductivity in monolayer h-B2O via doping in the diffusive regime. Sci Rep 16, 7679 (2026). https://doi.org/10.1038/s41598-026-38967-w
Palavras-chave: materiais bidimensionais, borofeno oxidado, condutividade térmica eletrônica, transporte de calor anisotrópico, controle por dopagem