Clear Sky Science · pt
Otimização computacional da solubilidade do domínio calpaína DEK1 por modelagem estrutural integrada e mutagênese direcionada baseada em dados
Por que fazer proteínas de plantas se comportarem importa
Muitas das proteínas que controlam como as plantas crescem são moléculas grandes e frágeis que não se dissolvem quando os cientistas tentam estudá-las em laboratório. Uma dessas proteínas, chamada DEK1, ajuda a moldar o corpo da planta desde o nível de células individuais. Mas, porque uma parte crucial do DEK1 tende a agregar quando produzida em bactérias, sua estrutura 3D permaneceu desconhecida, retardando esforços para entendê‑la e aproveitá‑la. Este estudo mostra como a modelagem por computador e um desenho inteligente baseado em dados podem redesenhar essa região problemática para torná‑la mais solúvel, sem comprometer sua arquitetura — oferecendo uma receita geral para domar proteínas difíceis.
Alvejando o ponto problemático em uma proteína vegetal chave
DEK1 é uma proteína incomumente grande, embutida nas membranas celulares e terminada por uma região enzimática de clivagem conhecida como domínio calpaína. Estudos genéticos mostraram que esse domínio é essencial para o desenvolvimento normal em plantas como musgos e culturas, contudo sua estrutura nunca foi resolvida experimentalmente. Quando pesquisadores tentam produzir esse núcleo calpaína (chamado CysPc) na bactéria hospedeira comum Escherichia coli, ele tende a tornar‑se insolúvel e formar corpos de inclusão densos. Isso torna praticamente impossível purificá‑lo nas quantidades e qualidade necessárias para estudos estruturais e funcionais detalhados. Os autores, portanto, propuseram redesenhar o domínio CysPc para que se dissolvesse com mais facilidade, preservando sua forma global.
Construindo um modelo 3D confiável do zero
Como não existe uma estrutura experimental para essa calpaína vegetal, a equipe primeiro precisou prever sua forma 3D. Eles combinaram várias ferramentas de predição estrutural de ponta, incluindo AlphaFold2, SWISS‑MODEL e I‑TASSER, e ancoraram essas predições em estruturas conhecidas de calpaínas mamíferas relacionadas. Usando uma abordagem de consenso, refinaram e checaram os modelos resultantes com múltiplos testes de qualidade que avaliam a geometria da espinha dorsal, o empacotamento e a conformidade com padrões estruturais conhecidos. Essas verificações independentes mostraram que o modelo integrado do domínio CysPc era mais confiável do que qualquer predição isolada, fornecendo um ponto de partida sólido para explorar como pequenas alterações na sequência de aminoácidos poderiam melhorar a solubilidade.
Testando mutações virtuais em um solvente simulado
Com o modelo 3D em mãos, os autores executaram extensas simulações de dinâmica molecular, nas quais a proteína e as moléculas de água circundantes são acompanhadas ao longo do tempo no computador. Eles focaram em resíduos na superfície da proteína que eram flexíveis, hidrofóbicos ou previstos como promotores de agregação. Posições candidatas foram mutadas individualmente para aminoácidos mais amigáveis à água e então simuladas por 200 nanosegundos cada. Para cada variante, mediram características relacionadas à solubilidade, como a área de superfície em contato com a água, quão compacta a proteína permanecia e a intensidade das flutuações atômicas. Muitas mutações únicas aumentaram modestamente a exposição ao solvente ou as ligações internas de hidrogênio sem alterar o dobramento geral, sugerindo que a estrutura básica do CysPc poderia tolerar substituições escolhidas com cuidado.
Deixando algoritmos vasculharem o espaço de mutações
Mudar apenas um resíduo raramente produz ganhos dramáticos em solubilidade, então os pesquisadores exploraram combinações de duas e três mutações. Eles geraram uma biblioteca de variantes duplas e triplas a partir das melhores mutações únicas e novamente simularam cada uma. Para pontuar e classificar esses desenhos de forma justa, definiram um índice ponderado que combina múltiplas características das simulações conhecidas por correlacionar com solubilidade, recompensando maior hidratação e ligação interna enquanto penaliza flexibilidade excessiva. Em seguida usaram um algoritmo de aprendizado por reforço (Proximal Policy Optimization) para navegar pelo enorme espaço de possíveis triplos mutantes e propor as combinações mais promissoras. Essa busca orientada por dados convergiu para um triplo mutante particular, chamado MUT347, como o principal candidato.
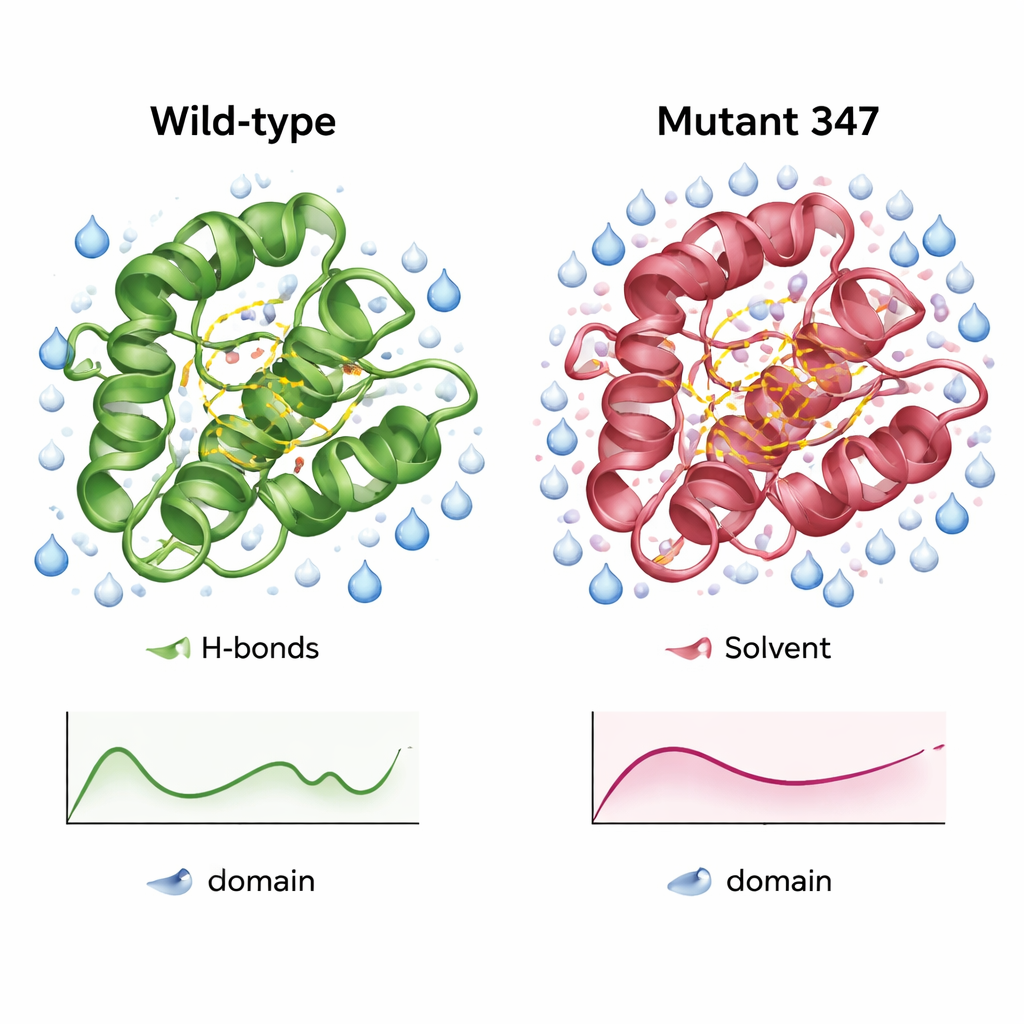
Uma versão mais compacta e melhor hidratada da enzima
Simulações detalhadas do domínio CysPc selvagem e do MUT347 revelaram como a variante projetada diferia. MUT347 equilibrou mais rapidamente e mostrou desvios gerais menores em relação à sua forma inicial, indicando maior estabilidade estrutural em solução. Suas alças e extremidades de cadeia estavam ligeiramente menos flácidas, enquanto a região catalítica central manteve sua flexibilidade original, sugerindo que movimentos importantes para a função foram preservados. O triplo mutante apresentou mais ligações internas de hidrogênio e uma maior superfície acessível à água em regiões-chave, sinais de uma superfície mais organizada e mais hidratada. Sob diferentes concentrações de sal e níveis de pH, MUT347 manteve consistentemente flutuações menores do que a proteína original, comportamento associado a menor tendência à agregação.
O que isso significa para estudar e reutilizar proteínas
Para não especialistas, a conclusão é que os autores construíram uma receita majoritariamente computacional para transformar um fragmento difícil e agregante de uma proteína vegetal vital em uma versão mais solúvel e comportada, sem precisar conhecer sua estrutura experimentalmente de antemão. Ao combinar predição moderna de estruturas, simulações em escalas de tempo longas e algoritmos de aprendizado capazes de equilibrar muitas escolhas de projeto simultaneamente, eles identificaram uma tripla mutação prevista para estabilizar o dobramento e expô‑lo de forma mais favorável à água. Embora trabalhos experimentais ainda sejam necessários para confirmar os ganhos em tubos de ensaio reais, essa estrutura analítica pode ser amplamente útil para resgatar outras proteínas eucarióticas difíceis de produzir, ajudando cientistas a desbloquear estruturas e funções atualmente fora de alcance.
Citação: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Palavras-chave: solubilidade de proteína, mutagênese computacional, dinâmica molecular, calpaína vegetal DEK1, engenharia de proteínas