Clear Sky Science · pt
Potencial interatômico por aprendizado de máquina para as propriedades estruturais dos óxidos de ferro
Por que as Pedras Enferrujadas Importam
Os óxidos de ferro – os minerais que dão à ferrugem sua cor – sustentam discretamente grande parte da vida moderna. São a principal fonte de ferro para o aço, ingredientes-chave em baterias e células solares e até ajudam a depurar água poluída. Ainda assim, apesar de sua importância, continuamos com dificuldades para prever como esses materiais se comportam em condições reais, especialmente ao nível atômico. Este artigo descreve como pesquisadores usaram inteligência artificial moderna para construir um modelo digital rápido e preciso de um óxido de ferro crucial, a hematita, abrindo caminho para experimentos virtuais mais confiáveis em tudo, desde beneficiamento de minério até dispositivos de energia limpa.
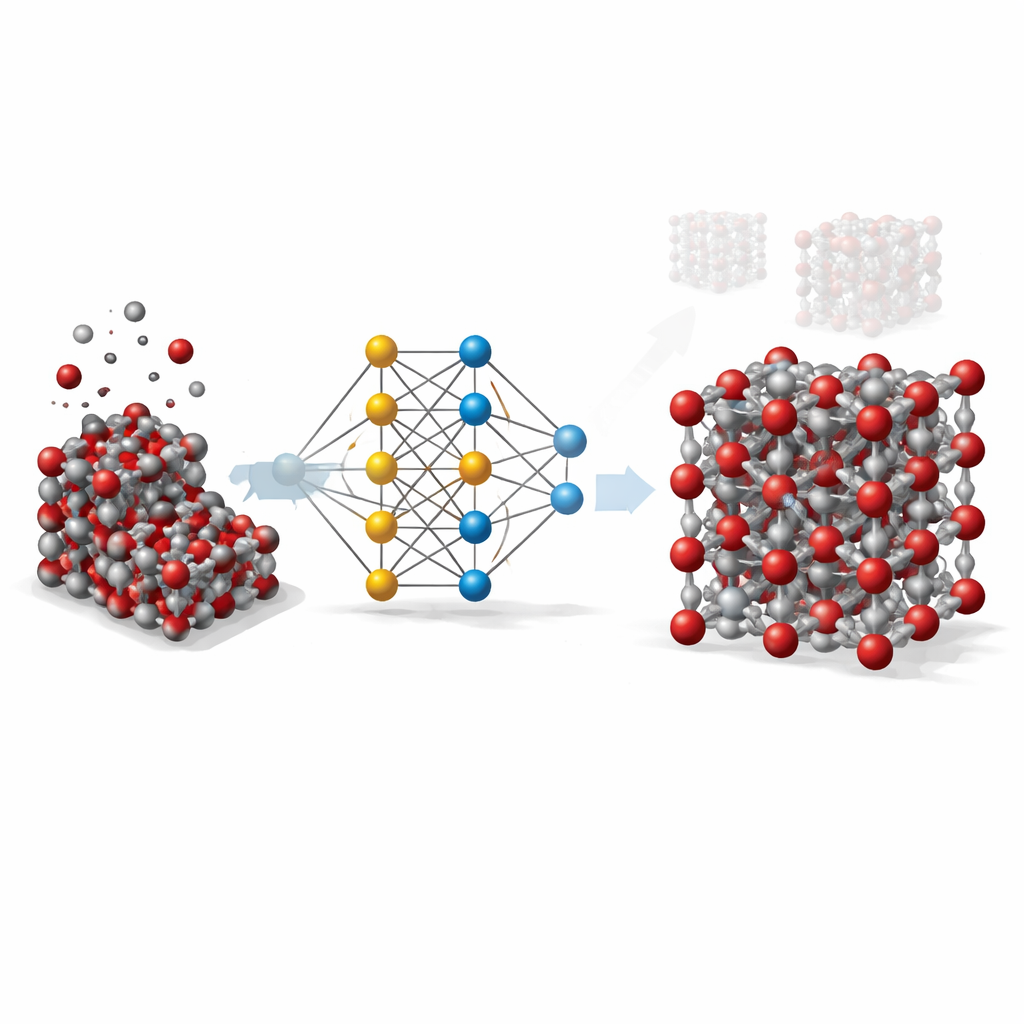
De Cálculos Caros a Atalhos Inteligentes
Para entender um sólido como a hematita em detalhe, os cientistas idealmente recorrem a métodos quântico-mecânicos que acompanham como elétrons e átomos interagem. Esses métodos, embora muito precisos, são tão exigentes computacionalmente que se tornam impraticáveis para simular grandes amostras ou longos intervalos de tempo. Modelos clássicos, em contraste, são rápidos porém rudimentares: dependem de fórmulas simples ajustadas para situações específicas e frequentemente falham quando temperatura, pressão ou forma do cristal mudam. O trabalho aqui apresentado busca preencher essa lacuna usando aprendizado de máquina para imitar a precisão dos cálculos quânticos ao mesmo tempo em que mantém a velocidade dos modelos tradicionais.
Ensinando uma Rede Neural sobre Átomos
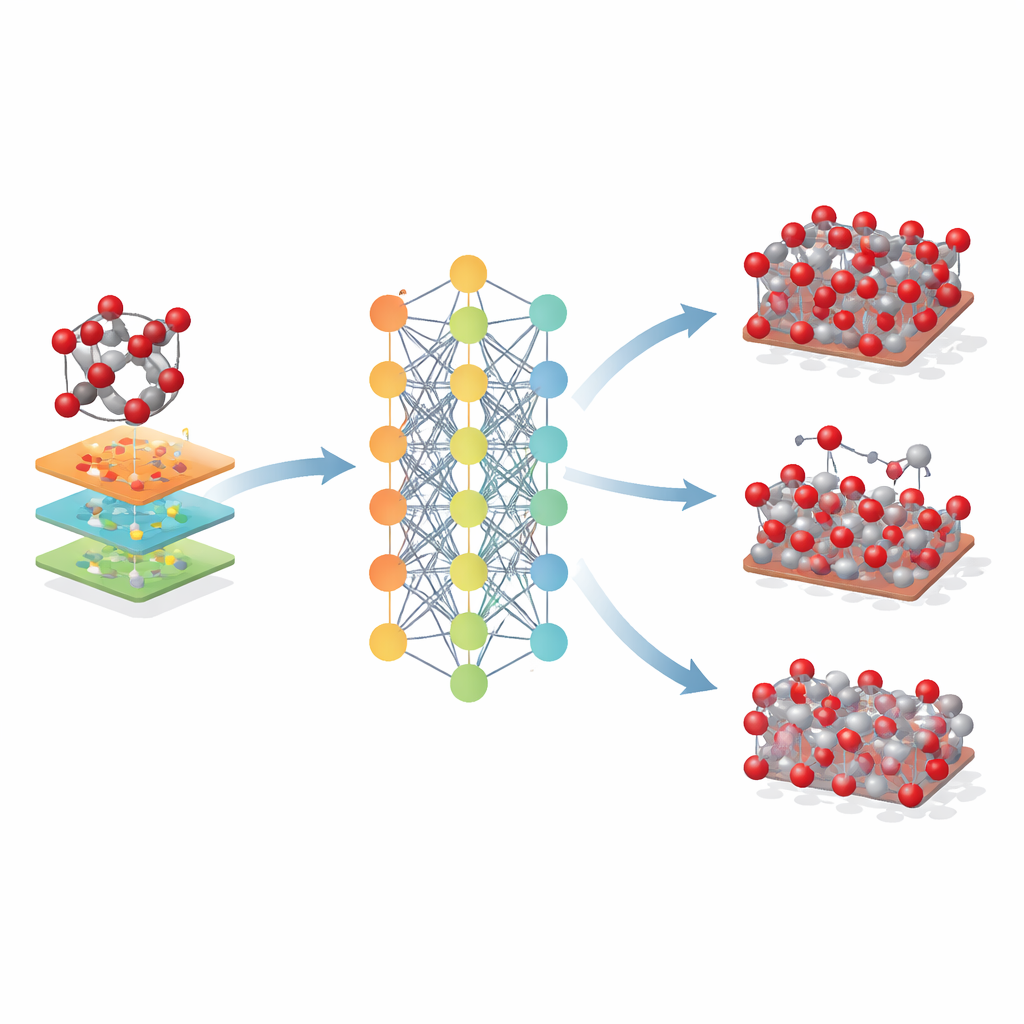
A equipe construiu o que se conhece como um potencial baseado em rede neural gráfica para a hematita. Nessa abordagem, cada átomo é tratado como um nó na rede, e as ligações e átomos vizinhos são as conexões entre os nós. Para ensinar essa rede como os átomos na hematita se atraem e se repelem, os pesquisadores primeiro geraram milhares de instantâneos atômicos usando simulações padrão ao longo de uma ampla gama de temperaturas, pressões e distorções cristalinas, incluindo tanto cristais no volume quanto superfícies expostas. Em seguida, usaram um método quântico de alto nível (DFT+U) para calcular a energia, as forças e as tensões internas de cada instantâneo, e treinaram a rede neural para reproduzir esses valores o mais fielmente possível.
Verificando o Modelo Contra a Realidade
Uma vez treinado, o novo potencial – chamado Fe-MLIP – foi rigorosamente testado. Os autores compararam suas previsões para quantidades estruturais básicas, como dimensões da rede e a forma como o cristal se deforma sob tensão, com experimentos e com vários modelos clássicos amplamente usados. O Fe-MLIP reproduziu a estrutura cristalina conhecida da hematita com erro de poucos por cento e capturou seu comportamento elástico quase tão bem quanto cálculos quânticos diretos, superando claramente outros campos de força para muitas propriedades. Também teve bom desempenho em testes mais sutis, como a expansão do material com a temperatura e as vibrações dos átomos, importantes para transporte de calor e espectroscopia. Essas frequências vibracionais, nunca mostradas explicitamente durante o treinamento, ficaram mais próximas dos valores medidos do que as obtidas por modelos concorrentes.
Indo Além de um Único Mineral
Os pesquisadores então exploraram até onde esse modelo baseado em hematita poderia ser estendido. Aplicaram-no a óxidos de ferro relacionados – maghemita e magnetita – que compartilham blocos atômicos semelhantes, mas diferem na organização cristalina e nos estados de oxidação do ferro. Mesmo não tendo sido treinado nessas fases, o Fe-MLIP produziu valores razoáveis para os tamanhos das redes e rigidez, frequentemente igualando ou superando modelos clássicos especializados. O potencial também capturou a estabilidade relativa de superfícies cristalinas chave e até tendências no custo energético de criar vacâncias atômicas, características cruciais para entender corrosão, catálise e desempenho de baterias.
O Que Isso Significa para o Projeto de Materiais Futuro
Para não especialistas, a conclusão é que este trabalho fornece um poderoso “gêmeo digital” para os óxidos de ferro. O modelo Fe-MLIP permite que pesquisadores executem simulações grandes e de longa duração da hematita e materiais relacionados com confiabilidade próxima ao nível quântico, mas a uma fração do custo. Embora herde algumas limitações do método quântico subjacente e esteja atualmente focado em ferro e oxigênio, já possibilita estudos mais realistas de como esses minerais respondem a tensão, calor, superfícies e defeitos. Em termos práticos, tal ferramenta pode acelerar o desenvolvimento de processos de siderurgia melhores, catalisadores e baterias mais eficientes e tecnologias ambientais aprimoradas que dependem de óxidos de ferro – tudo ao permitir que cientistas testem ideias no computador antes de irem ao laboratório ou à mina.
Citação: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Palavras-chave: hematita, óxidos de ferro, potencial por aprendizado de máquina, redes neurais gráficas, dinâmica molecular