Clear Sky Science · pt
Otimização da seleção de características em dados de microarranjos de câncer usando uma estrutura evolutiva baseada em heap para espaços de alta dimensão
Por que escolher os genes certos importa
Testes de câncer construídos a partir de tecnologias genéticas modernas podem medir dezenas de milhares de genes de uma só vez, mas os médicos frequentemente dispõem de dados de apenas algumas dezenas de pacientes. Ocultos nessa enorme “selva gênica” estão um número muito menor de sinais que realmente diferenciam um tipo de câncer de outro, ou um tumor de tecido saudável. Este artigo apresenta um novo método de busca inteligente para identificar automaticamente esses genes-chave, com o objetivo de tornar o diagnóstico assistido por computador mais preciso, rápido e mais fácil de interpretar.
Demasiados sinais, poucos dados
Experimentos com microarranjos e tecnologias semelhantes permitem aos pesquisadores medir níveis de atividade de milhares de genes em cada amostra de paciente. Ainda assim, o número de amostras costuma ser muito pequeno, às vezes inferior a cem. Muitas dessas leituras gênicas são ruidosas, redundantes ou irrelevantes para a doença em questão. Manter todas elas pode sobrecarregar algoritmos de aprendizado, desacelerar o processamento e produzir modelos enganosos que se apegam a peculiaridades aleatórias em vez de biologia verdadeira. O processo de reduzir esse conjunto a um subconjunto útil é chamado de “seleção de características”, e é crucial se quisermos previsões confiáveis a partir de dados médicos de alta dimensionalidade.
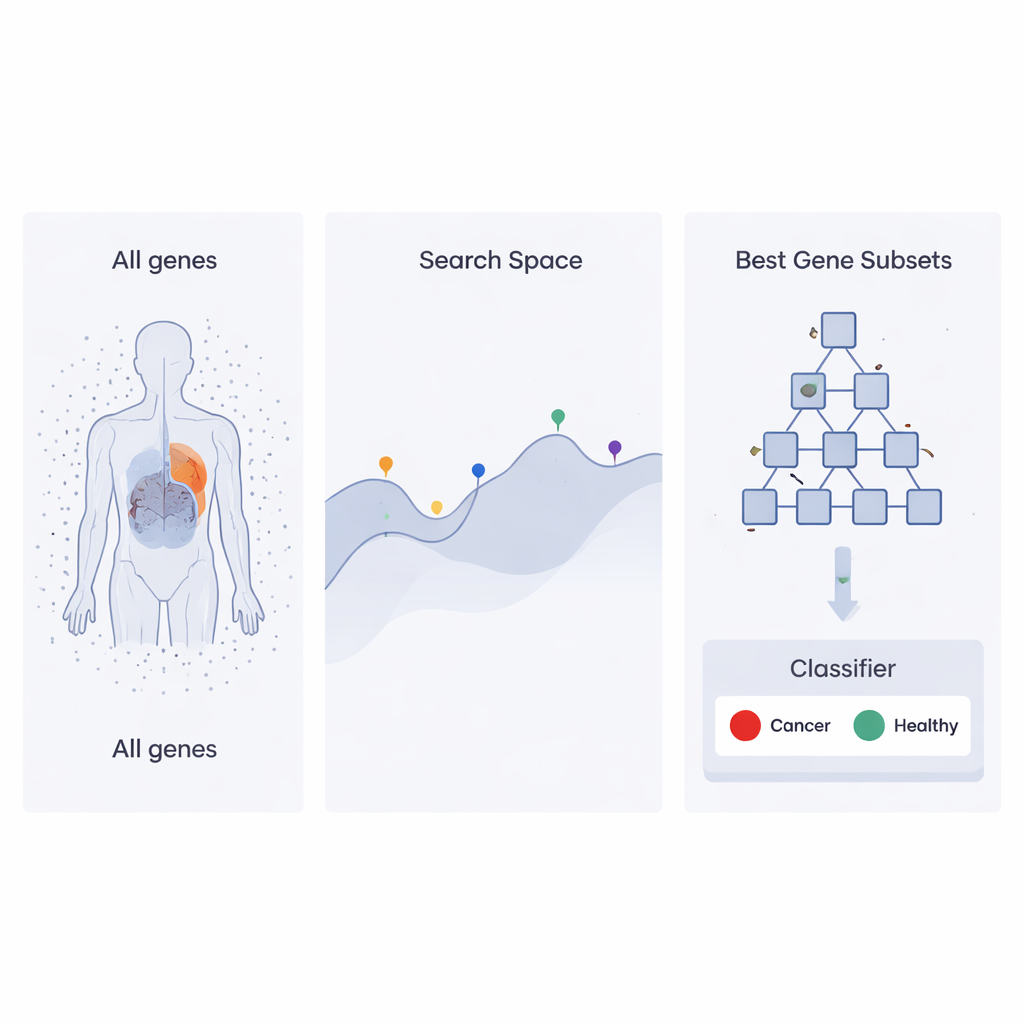
Uma estratégia de busca inspirada em hierarquias corporativas
Os autores constroem sobre uma abordagem de otimização recente chamada Heap‑Based Optimizer (HBO), que toma ideias da forma como funcionários são organizados em uma empresa. Imagine cada conjunto possível de genes como um “funcionário” cujo desempenho é avaliado por quão bem ele ajuda um classificador a distinguir amostras de câncer de amostras saudáveis. Esses “funcionários” são organizados em uma hierarquia, como uma escada corporativa, usando uma estrutura de dados conhecida como heap. Conjuntos de genes com bom desempenho ficam próximos ao topo, enquanto os mais fracos ficam mais abaixo. Ao longo de muitas rodadas, os empregados de menor posição ajustam suas escolhas copiando e modificando levemente o que seus superiores e colegas fazem, empurrando gradualmente toda a organização em direção a soluções melhores.
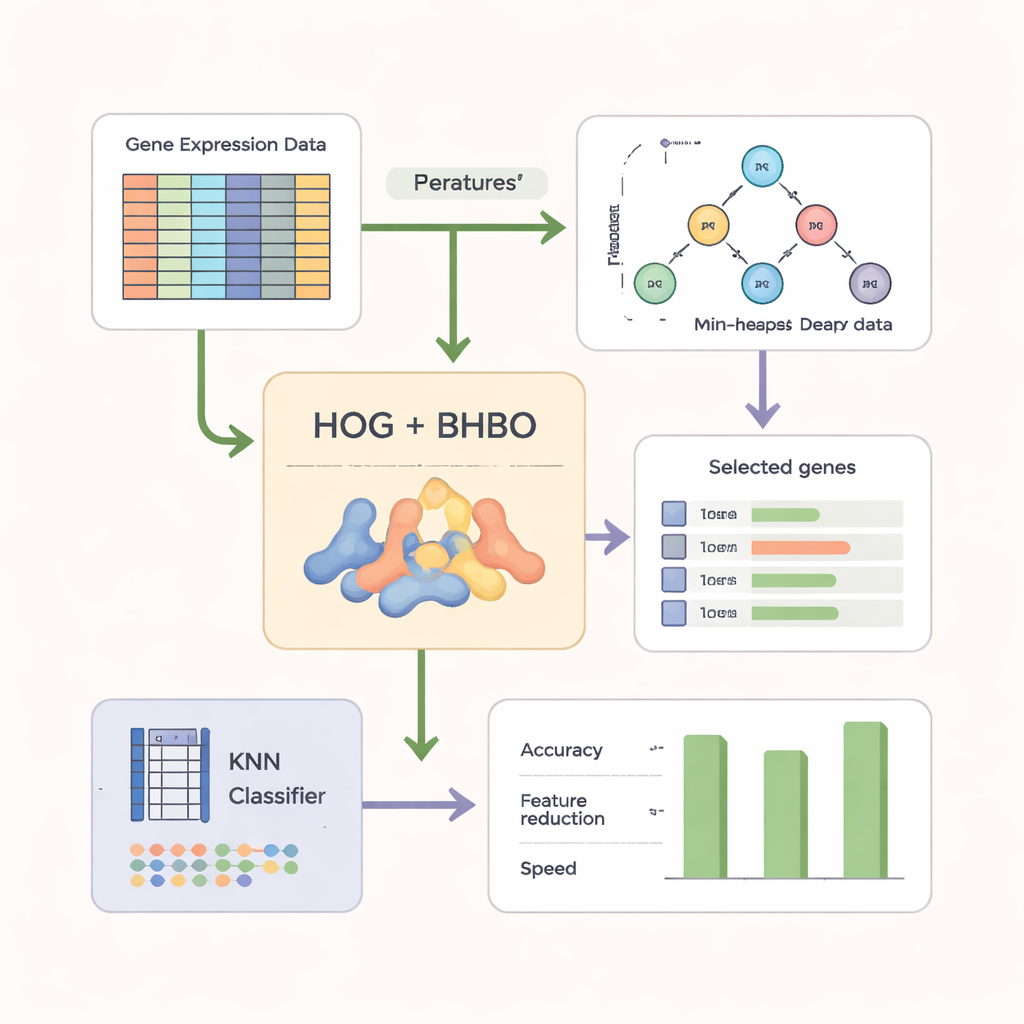
Transformando dados gênicos brutos em padrões mais nítidos
Para tornar a busca mais eficaz, os autores não se apoiam apenas nas leituras gênicas brutas. Primeiro, eles remodelam os dados de microarranjo em uma forma semelhante a imagem e aplicam uma técnica chamada Histogram of Oriented Gradients (HOG), amplamente usada em visão computacional. O HOG captura como os níveis de expressão variam ao longo dos genes, destacando padrões locais em vez de medições isoladas. Essas características baseadas em padrão são então combinadas com as informações gênicas originais. Um classificador simples chamado k‑Nearest Neighbors (KNN) serve como “juiz”, pontuando cada subconjunto de genes candidato pela precisão com que rotula novas amostras, ao mesmo tempo em que recompensa conjuntos menores e mais compactos.
Testes em múltiplos conjuntos de dados de câncer
Os pesquisadores avaliaram sua versão binária do Heap‑Based Optimizer (BHBO) em nove conjuntos de dados públicos de microarranjos de câncer, incluindo tumores cerebrais, leucemias, câncer de próstata e coleções mistas de tumores com muitos subtipos. Cada conjunto tinha milhares até mais de quinze mil genes medidos, mas relativamente poucas amostras de pacientes. Para cada conjunto, o BHBO foi executado repetidamente e comparado com sete métodos de busca bem conhecidos, como algoritmos genéticos e otimização por enxame de partículas. A equipe mediu não apenas a acurácia, mas também quantos genes foram mantidos, com que rapidez a busca convergia e quão estáveis eram os resultados quando os dados eram perturbados por ruído simulado, efeitos de lote e erros de rotulagem.
O que o novo método alcançou
Ao longo dos nove conjuntos, a abordagem baseada em heap alcançou uma acurácia média de classificação de cerca de 95% enquanto reduzia o número de genes em mais de 85%. Ela superou claramente métodos concorrentes em vários conjuntos e mostrou convergência mais rápida, ou seja, encontrou bons conjuntos de genes em menos passos de busca. Mesmo quando os autores deliberadamente corromperam os dados — adicionando ruído ou invertendo alguns rótulos de amostra — o desempenho do método caiu apenas ligeiramente e permaneceu superior às alternativas. Testes estatísticos confirmaram que esses ganhos eram improváveis de ocorrer por acaso.
O que isso significa para futuros diagnósticos de câncer
Na prática, este trabalho mostra que uma estratégia de busca cuidadosamente projetada pode peneirar enormes conjuntos genéticos e descobrir pequenos painéis de genes ricos em informação que ainda classificam cânceres muito bem. Para clínicos e pesquisadores, conjuntos gênicos compactos são mais fáceis de validar biologicamente, mais baratos de medir em testes de acompanhamento e mais adequados para integração em ferramentas de suporte à decisão. Embora o método não descubra diretamente novos medicamentos ou vias, ele focaliza melhor marcadores genéticos promissores, ajudando outros estudos a se concentrarem nos sinais mais informativos ocultos em dados de câncer de alta dimensionalidade.
Citação: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Palavras-chave: microarranjo de câncer, seleção de características, otimização metaheurística, biomarcadores genéticos, mineração de dados médicos