Clear Sky Science · pt
Um método para detecção de variantes estruturais usando matriz de contatos Hi-C e redes neurais
Por que dobrar o DNA em 3D importa
Nosso DNA costuma ser representado como uma simples cadeia de letras, mas dentro de cada célula ele se dobra em uma forma tridimensional complexa. Quando grandes trechos dessa cadeia são deletados, invertidos ou deslocados — alterações chamadas de variações estruturais — eles podem interromper genes e contribuir para o desenvolvimento do câncer. Este estudo apresenta o VarHiCNet, um novo sistema de inteligência artificial que lê mapas de dobramento 3D do DNA e identifica essas alterações em grande escala com mais precisão do que ferramentas existentes, oferecendo uma nova maneira de estudar genomas de câncer e outras doenças.
Vendo mudanças no genoma através de mapas de contato 3D
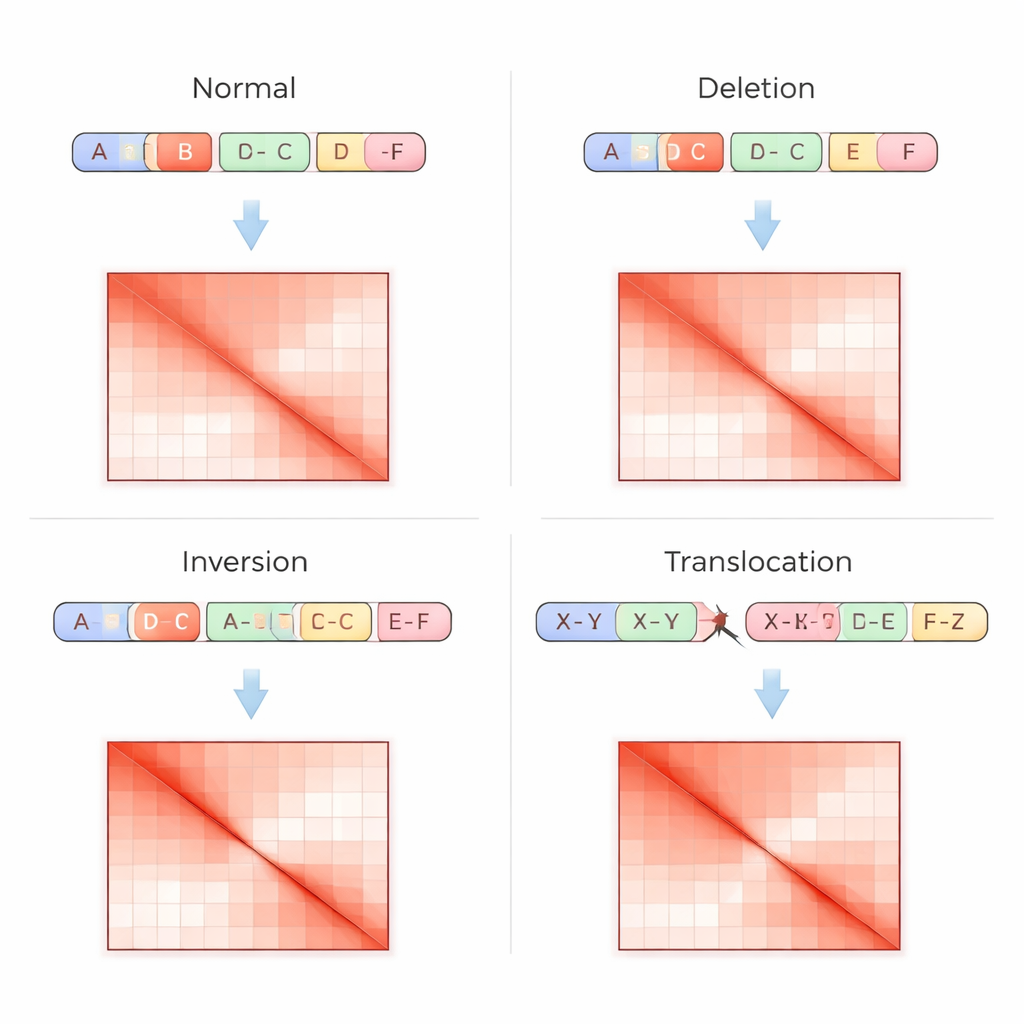
Testes genômicos tradicionais leem o DNA como uma sequência linear, o que dificulta a detecção de rearranjos complexos, especialmente em regiões repetitivas ou quando trechos são movidos sem alterar seu número de cópias. A técnica Hi-C aborda o problema de forma diferente: ela mede com que frequência partes distantes do DNA tocam-se fisicamente dentro do núcleo e registra esses contatos em uma grade, ou matriz de contatos, onde pontos mais brilhantes indicam interação mais forte. Variações estruturais deixam impressões digitais distintivas nessas matrizes — como faixas ausentes quando uma região foi deletada, padrões espelhados quando um segmento é invertido, ou pontos fora da diagonal quando dois cromossomos se fundem. O VarHiCNet foi projetado para reconhecer automaticamente esses padrões visuais.
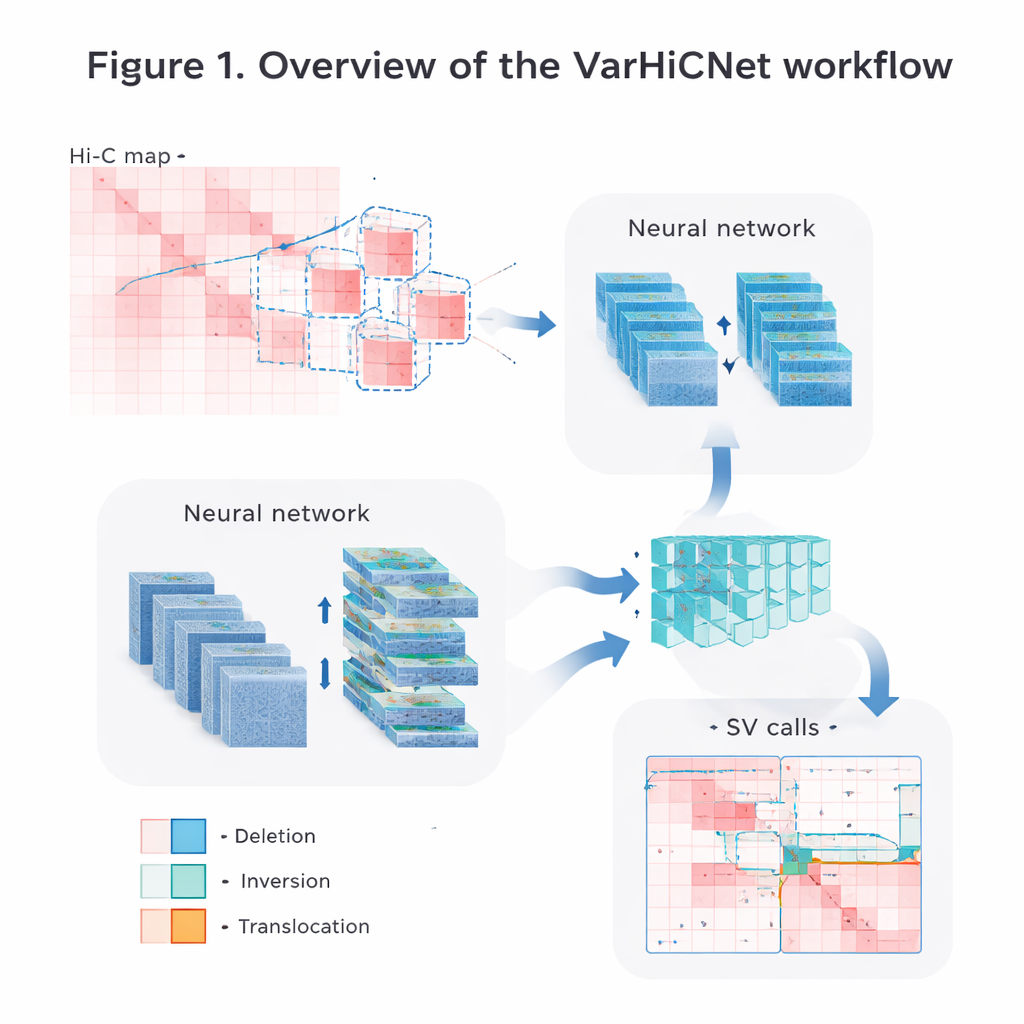
Transformando mapas genômicos em imagens para IA
Os autores convertem os dados brutos de contato Hi-C em imagens que sistemas de visão computacional podem processar facilmente. Primeiro, eles normalizam cuidadosamente as matrizes para corrigir a queda natural na frequência de contato à medida que segmentos de DNA se afastam, preservando sinais de interação tanto de curto quanto de longo alcance. Em seguida, escaneiam cada cromossomo com janelas quadradas sobrepostas e recortam muitas submatrizes menores. Cada submatriz é redimensionada para uma imagem colorida padronizada de 800 por 800 pixels, onde diferentes intensidades de contato são mapeadas em tons avermelhados pelos três canais de cor. Essa representação semelhante a imagem permite ao modelo reaproveitar técnicas poderosas originalmente desenvolvidas para reconhecer objetos em fotografias.
Emprestando truques da detecção de objetos
O VarHiCNet trata cada variante estrutural potencial como se fosse um “objeto” em uma imagem. Ele se baseia em um moderno framework de detecção de objetos chamado RT-DETR, que usa uma combinação de redes neurais convolucionais e Transformers para realçar regiões importantes. Uma espinha dorsal ResNet primeiro extrai características em múltiplas escalas: camadas rasas preservam os detalhes finos necessários para localizar pontos de quebra exatos, enquanto camadas mais profundas capturam padrões mais amplos que sinalizam eventos grandes. Um módulo de fusão de características então mistura informações de várias camadas para que pistas locais e globais sejam preservadas. Outro bloco personalizado, inspirado em pooling piramidal espacial, ajusta quanto da região circundante o modelo “vê” de uma vez, tornando-o sensível a variantes que se estendem desde trechos relativamente pequenos até intervalos muito grandes do DNA.
De regiões candidatas a tipos precisos de variante
Uma vez que o VarHiCNet propõe regiões candidatas na imagem Hi-C, ele precisa refiná-las em pontos de quebra exatos e tipos específicos de variante, como deleções, inversões, duplicações ou translocações. Para isso, o sistema amplia a vizinhança em torno de cada ponto de quebra previsto e reduz sua complexidade usando uma técnica matemática chamada análise de componentes principais, que destaca onde o padrão de contatos muda mais abruptamente. Essas representações compactas são então alimentadas em um classificador baseado em Transformer que aprende diferenças sutis nos padrões locais para cada categoria de variante. O resultado é uma chamada detalhada para cada evento: onde ele ocorre no genoma e que tipo de alteração estrutural representa.
Desempenho em diferentes linhagens celulares de câncer
Os pesquisadores testaram o VarHiCNet em dados Hi-C de seis diferentes linhagens celulares humanas de câncer, cobrindo tumores de sangue, mama, cérebro, rim, pulmão e próstata. Usando um catálogo de alta confiança de variantes estruturais conhecidas como padrão-ouro, eles compararam seu método com várias ferramentas líderes que também analisam dados Hi-C. Em eventos tanto dentro quanto entre cromossomos, o VarHiCNet geralmente atingiu pontuações F1 mais altas ou comparáveis, o que significa que equilibrou sensibilidade e precisão melhor do que outras abordagens. Foi particularmente eficaz na detecção de translocações balanceadas e inversões — rearranjos que muitas vezes deixam pouca evidência em sequenciamento de DNA padrão, mas deixam assinaturas claras no dobramento 3D. Os autores também mostraram que suas escolhas de projeto, como a resolução da imagem e os módulos de fusão de características, melhoraram consistentemente o desempenho em testes controlados.
O que isso significa para entender doenças
Em termos práticos, o VarHiCNet oferece aos cientistas uma maneira mais inteligente de “ver” como o genoma se dobra em 3D e de identificar grandes rearranjos relacionados a doenças que podem passar despercebidos pelo sequenciamento convencional. Ao transformar mapas de contato complexos em imagens e aplicar redes neurais modernas no estilo de visão computacional, o método pode detectar e categorizar muitos tipos de variações estruturais com alta confiabilidade em diferentes tipos de câncer. Embora ainda tenha dificuldades com algumas alterações muito pequenas ou altamente complexas e dependa de dados de treinamento ricos, o VarHiCNet aponta para um futuro em que a arquitetura 3D do genoma se torne parte rotineira de como lemos, interpretamos e, eventualmente, direcionamos as mudanças genéticas que sustentam o câncer e outras doenças.
Citação: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Palavras-chave: variação estrutural, Hi-C, aprendizado profundo, genômica do câncer, genoma 3D