Clear Sky Science · pt
Mutação GJB2 c.109G > A ativa via mitocondrial IFI27 que leva à apoptose e surdez não sindrômica hereditária
Por que células minúsculas do ouvido importam para o futuro das crianças
A perda auditiva presente ao nascer afeta milhões de crianças no mundo, moldando muitas vezes como elas aprendem a falar, têm sucesso na escola e se relacionam. Um dos culpados genéticos mais comuns é um gene chamado GJB2, mas os médicos ainda não compreendiam totalmente como alterações nesse gene danificam o ouvido interno. Este estudo usa zebrafish e células humanas para traçar a cadeia de eventos desde uma única mudança no DNA do GJB2 até a morte das delicadas células sensoriais do som, e aponta uma nova molécula, IFI27, como possível alvo para tratamentos futuros.
Uma alteração genética comum por trás do silêncio infantil
Os pesquisadores começaram triando amostras de sangue de 1.199 crianças com suspeita de perda auditiva hereditária na província de Fujian, China. Eles concentraram-se em vários genes de surdez bem conhecidos e descobriram que alterações em GJB2 dominavam o quadro, representando 85% de todas as mutações detectadas. Entre elas, uma mudança específica chamada c.109G>A (também conhecida como p.Val37Ile) foi a mais frequente. Essa variante é relativamente comum na população geral, mas fortemente enriquecida em pessoas com perda auditiva, sugerindo que desempenha papel importante na perda auditiva não sindrômica — problemas auditivos que ocorrem sem outras manifestações médicas.
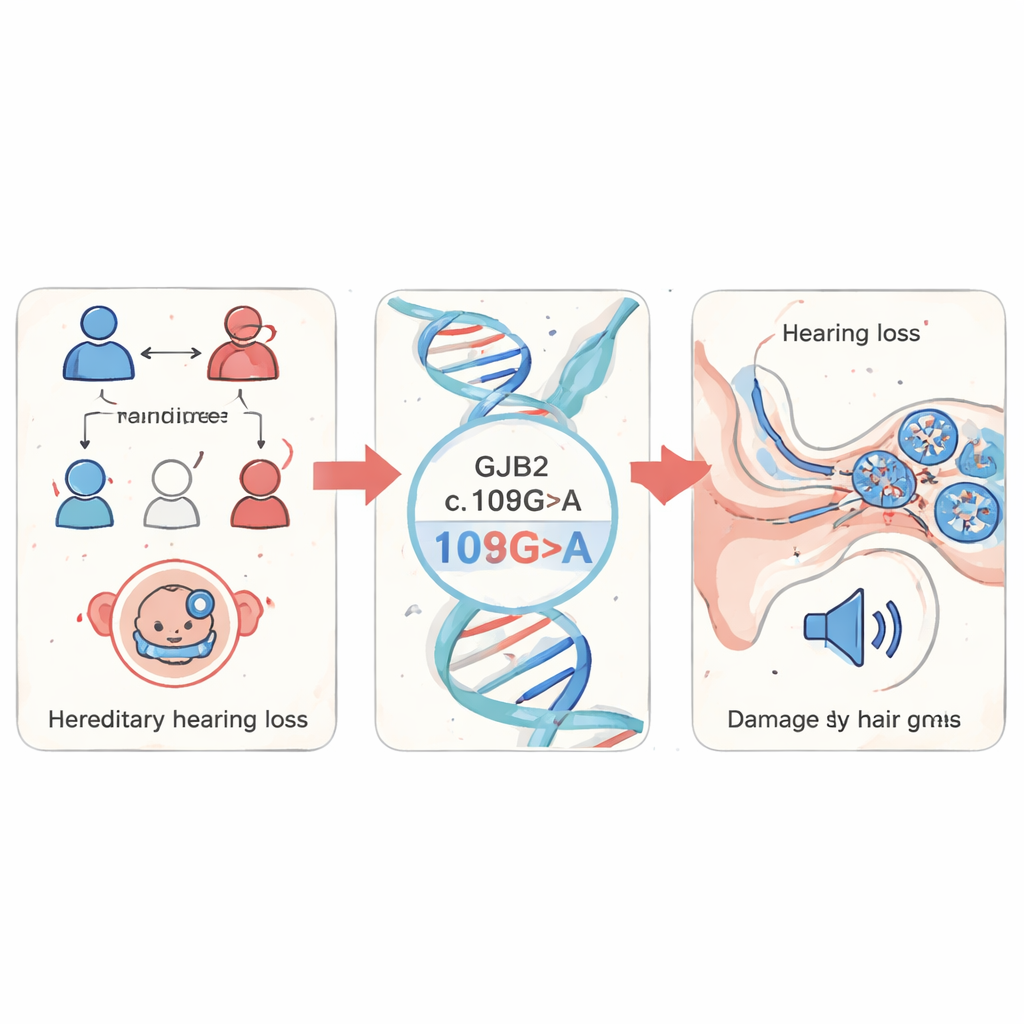
Seguindo o dano em um peixe transparente
Para ver o que essa mutação faz em um organismo vivo, a equipe recorreu ao zebrafish, um pequeno peixe de água doce cujos embriões são transparentes e compartilham muitos genes e estruturas do ouvido com os humanos. Eles engenheiraram embriões de zebrafish para produzir GJB2 humana normal ou a versão mutante c.109G>A, e também usaram uma abordagem de “knockdown” para reduzir o próprio gene gjb2 do peixe. Embriões com o gene mutante ou reduzido apresentaram crescimento retardado, cauda curvada e inchaço ao redor do coração, sinais de desenvolvimento comprometido. Mais importante, seus ouvidos internos eram claramente anormais: estruturas-chave chamadas otólitos estavam menores e mais afastadas, e a região cochlear preenchida por fluido estava reduzida. Quando os cientistas reintroduziram GJB2 normal junto com a mutante, muitos desses problemas estruturais melhoraram, mostrando que a mutação em si conduzia os defeitos.
De ouvidos defeituosos a comportamento auditivo prejudicado
Como a audição depende de minúsculas “células ciliadas” que convertem vibrações sonoras em sinais nervosos, a equipe corou essas células no zebrafish. Peixes com a mutação GJB2 ou com knockdown apresentaram muito menos células ciliadas tanto no ouvido interno quanto ao longo da superfície corporal, onde os zebrafish também detectam movimentação da água. Em seguida, os pesquisadores testaram quão bem os peixes respondiam a som. Usando um sistema automatizado de rastreamento, mediram distância e velocidade com que larvas de 5 dias nadavam quando expostas a rajadas breves de som. Peixes normais e com GJB2 selvagem reagiram nadando mais e mais rápido, enquanto peixes mutantes e com knockdown mal mudaram o comportamento, indicando audição prejudicada. Novamente, a adição de GJB2 normal restaurou em parte tanto o número de células ciliadas quanto o movimento induzido pelo som.
Um caminho de morte dentro das usinas da célula
Para entender o que ocorria dentro das células, os cientistas usaram sequenciamento de RNA para comparar a atividade gênica entre zebrafish normais e aqueles com gjb2 reduzido. Um conjunto de genes ligado à “via de apoptose mitocondrial” — uma rota de autodestruição centrada nas usinas de energia da célula — foi fortemente ativado. Em particular, vários membros da família IFI27 se destacaram, junto com conhecidos mediadores da morte celular como Bax, citocromo c, Apaf1 e caspases. Experimentos complementares em células HEK293 humanas confirmaram o padrão: células com o GJB2 mutante produziram mais espécies reativas de oxigênio (ROS, uma forma de estresse oxidativo), liberaram mais citocromo c das mitocôndrias e ativaram proteínas da apoptose, resultando em maior morte celular. Quando os pesquisadores silenciaram IFI27 em células com o gene mutante, os níveis de ROS diminuíram, os sinais de morte foram atenuados e menos células passaram por apoptose.
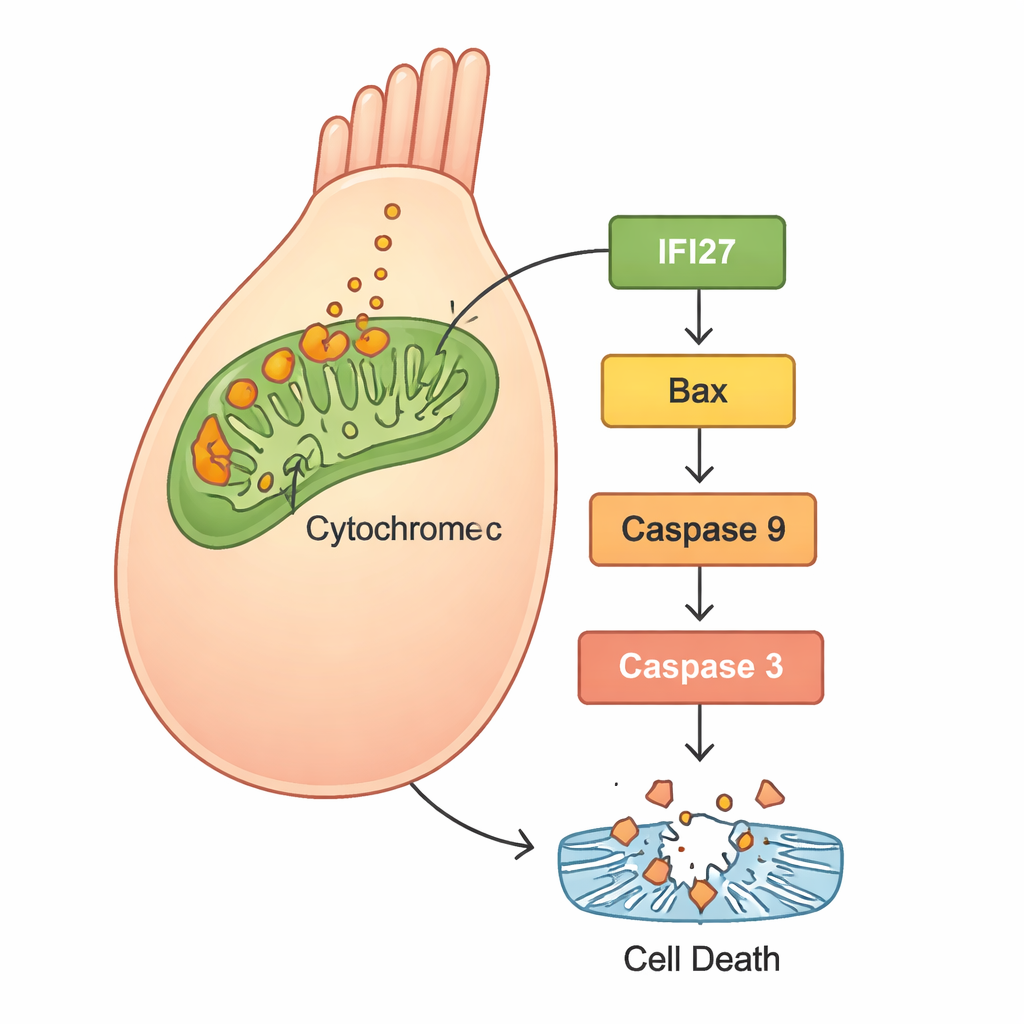
O que isso significa para tratamentos futuros
Em conjunto, os achados sugerem uma história clara: a mutação GJB2 c.109G>A perturba o desenvolvimento e a função normais do ouvido interno, não apenas alterando a comunicação celular, mas também desencadeando estresse nas mitocôndrias. Esse estresse eleva IFI27 e genes relacionados, libera citocromo c e ativa uma cascata de proteínas que empurram as células ciliadas para a morte programada. Como as células ciliadas não se regeneram facilmente em humanos, sua perda leva a déficits auditivos permanentes. Ao mostrar que reduzir IFI27 pode suavizar essa cascata destrutiva em células humanas, o estudo destaca IFI27 como um alvo promissor para fármacos ou terapias gênicas. Embora tais tratamentos ainda estejam distantes — e provavelmente precisarão ser administrados muito cedo na vida — este trabalho oferece um roteiro molecular concreto para transformar uma mutação genética antes misteriosa em uma causa potencialmente evitável de surdez infantil.
Citação: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Palavras-chave: surdez genética, mutação GJB2, modelo em zebrafish, apoptose mitocondrial, IFI27