Clear Sky Science · pt
Seleção de características simples, rápida e eficiente guiada por cluster fuzzy adaptativo para dados microarray bioinformáticos binários de alta dimensionalidade e fortemente desbalanceados
Por que isso importa para a pesquisa genética
Testes modernos de expressão gênica podem medir dezenas de milhares de genes em uma única amostra de paciente. Esse volume de dados promete diagnóstico de câncer mais precoce e escolhas de tratamento melhores, mas também cria um problema: a maioria desses genes é ruidosa, redundante ou está relacionada principalmente a casos comuns, não aos raros e perigosos. Este artigo apresenta uma nova forma de peneirar conjuntos massivos de dados de expressão gênica para que computadores possam identificar de maneira confiável pacientes pertencentes a um pequeno grupo minoritário difícil de detectar usando apenas um conjunto reduzido e cuidadosamente escolhido de genes.
O desafio de muitos genes muito semelhantes
Experimentos de microarray frequentemente acompanham milhares de níveis de atividade gênica para apenas algumas centenas de pacientes. Normalmente, uma classe (como um subtipo comum de câncer) supera em número a outra, criando dados fortemente desbalanceados. Nesse cenário, muitos genes se comportam de forma muito semelhante, e os padrões para pacientes da maioria e da minoria podem se sobrepor. Métodos de aprendizado padrão tendem a favorecer a classe majoritária e a se confundir com genes redundantes, o que leva a overfitting e a uma detecção ruim de subtipos raros. Métodos tradicionais de redução de dimensionalidade ou perdem interpretabilidade ao construir novas características mistas, ou selecionam genes sem avaliar detalhadamente o quanto eles ajudam um classificador a reconhecer os casos minoritários.
Um novo roteiro para seleção mais inteligente de genes
Os autores apresentam o AFCG‑SFE, um modelo adaptativo de seleção de características projetado especificamente para dados de expressão gênica de alta dimensionalidade e desbalanceados. O método parte de uma busca simples “de agente único” que liga ou desliga genes e testa quão bem eles suportam a classificação, mas o enriquece com várias etapas guiadas pelos dados. Primeiro, agrupa genes com base em como eles se comportam de forma similar, depois permite que genes pertençam a mais de um grupo para refletir a realidade biológica de que um gene pode estar envolvido em várias vias. Dentro de cada grupo, classifica os genes por quão informativos são sobre o rótulo da doença e mantém apenas alguns representantes-chave, reduzindo drasticamente a redundância antes mesmo do início da busca principal. 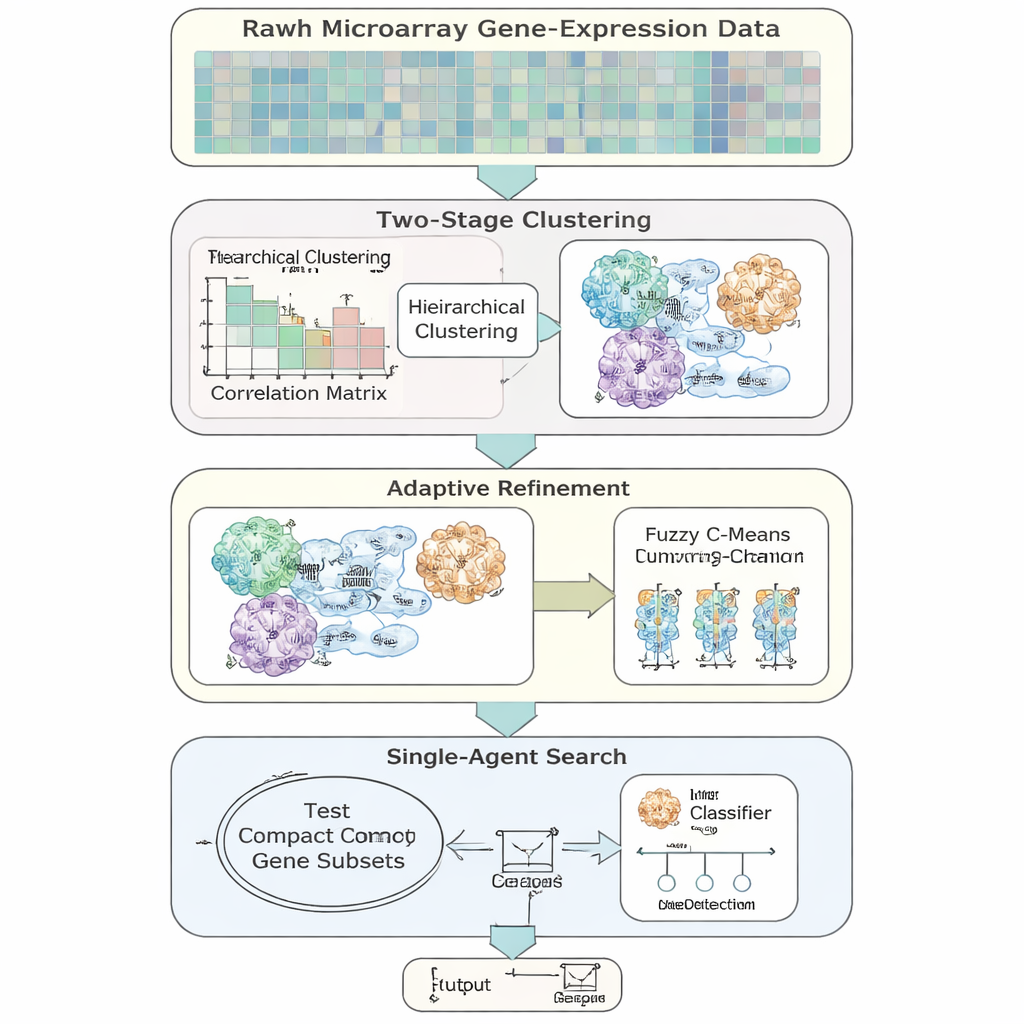
Fazer o computador se importar com pacientes raros
Em vez de focar na acurácia simples, o AFCG‑SFE usa uma função de aptidão que enfatiza métricas adequadas para dados enviesados, incluindo o equilíbrio entre identificar corretamente casos minoritários e majoritários e o desempenho ao longo de todos os limiares de decisão. A função de aptidão também inclui penalidades por selecionar muitos genes ou muitos genes do mesmo cluster, e uma recompensa para genes que apresentem forte dependência com o rótulo da doença. Importante, a intensidade dessas penalidades e recompensas é definida automaticamente a partir de propriedades do conjunto de dados, como quantos genes há por paciente e quanto as classes se sobrepõem, em vez de ajuste manual. Isso torna o método mais robusto e mais fácil de transferir entre estudos.
Adaptando-se à dificuldade do problema
Uma ideia-chave é que o algoritmo não deve sempre visar o menor conjunto de genes possível. Quando as duas classes são muito difíceis de separar ou estão fortemente sobrepostas, o método eleva automaticamente um limite inferior sobre quantos genes devem ser mantidos, garantindo que sinais raros, mas importantes, não sejam descartados. À medida que a busca progride, o AFCG‑SFE gradualmente aperta um limite por cluster sobre quantos genes podem sobreviver em cada grupo, enquanto ainda respeita esse mínimo. O resultado é um painel compacto e diverso de genes que captura a estrutura dos dados sem ser dominado por um único padrão redundante. 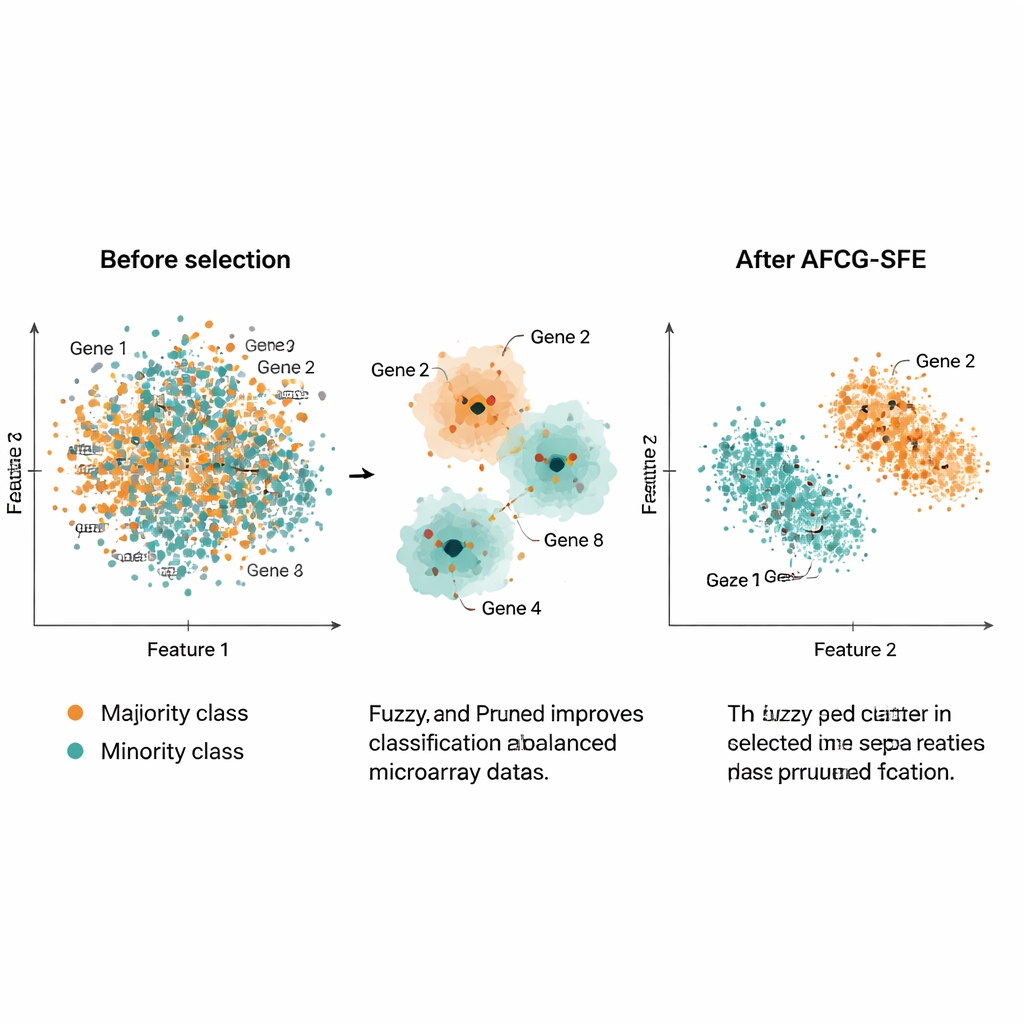
O que os experimentos mostram
Os autores testaram o AFCG‑SFE em 20 conjuntos públicos de microarray de câncer, cada um com milhares de genes, mas apenas cerca de 100–200 amostras e forte desbalanceamento de classes. Eles compararam o método com vários referenciais de busca evolutiva, filtros simples e abordagens incorporadas que integram seleção de características ao classificador. Em uma bateria de medidas — incluindo F‑measure, acurácia balanceada, área sob a curva ROC e uma medida de overfitting — o AFCG‑SFE foi o melhor ou empatou como melhor em todos os conjuntos de dados. Tipicamente selecionou menos de 25 genes (muitas vezes tão poucos quanto 6–8), removendo mais de 99% das características originais enquanto ainda melhorava ou mantinha o desempenho de classificação. Também reduziu um índice de complexidade que captura o quanto as classes se sobrepõem no espaço de características, indicando separação mais clara após a seleção.
Conclusão para não especialistas
Em termos práticos, este trabalho oferece uma forma de reduzir perfis enormes e ruidosos de expressão gênica a conjuntos muito pequenos de genes informativos que ainda permitem que computadores reconheçam com precisão subgrupos raros de pacientes. Ao agrupar inteligentemente genes semelhantes, recompensar aqueles que realmente acompanham a doença e proteger explicitamente contra vieses em favor da classe majoritária, o AFCG‑SFE entrega tanto previsões melhores quanto painéis de genes muito mais simples. Essa combinação pode ajudar pesquisadores a identificar potenciais biomarcadores, projetar testes diagnósticos mais interpretáveis e, em última instância, melhorar como ferramentas de medicina de precisão funcionam com dados biológicos reais e imperfeitos.
Citação: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Palavras-chave: expressão gênica, seleção de características, dados desbalanceados, microarray, subtipos de câncer