Clear Sky Science · pt
rhinotypeR permite atribuição reprodutível de genótipos de rinovírus a partir de sequências VP4/2
Por que os minúsculos vírus do resfriado ainda importam
A maioria de nós enxerga o resfriado comum como um incômodo, e não como uma ameaça séria. No entanto, os vírus que provocam muitos resfriados — os rinovírus humanos — também estão associados a infecções pulmonares graves, crises de asma e exacerbações de doenças pulmonares crônicas. Para acompanhar como esses vírus evoluem e se espalham, os cientistas precisam classificá‑los em “tipos” genéticos precisos, de modo semelhante a etiquetar produtos com códigos de barras. Este artigo apresenta o rhinotypeR, um pacote de software gratuito e de código aberto que torna essa rotulagem genética mais precisa, consistente e fácil de reproduzir, ajudando equipes de saúde pública a monitorar com mais clareza uma família de vírus respiratórios frequentemente subestimada.
A variedade oculta nos resfriados comuns
Os rinovírus humanos são extraordinariamente comuns, aparecendo em até 60% das amostras de pessoas com doença respiratória aguda. Longe de ser um único vírus, eles se dividem em três grupos principais, chamados A, B e C, e em pelo menos 169 tipos genéticos reconhecidos. Diferentes tipos se comportam de modos distintos: alguns estão mais frequentemente ligados a infecções graves em crianças e a crises de asma, enquanto outros aparecem com menos frequência em doenças sérias. Como esses tipos evoluem de forma independente e carregam características de superfície distintas, os cientistas precisam de maneiras confiáveis de distingui‑los para acompanhar como surtos se movem por escolas, lares e comunidades.
De ferramentas dispersas a um caminho claro
Até agora, atribuir um tipo de rinovírus a partir do seu código genético tem sido um trabalho fragmentado. Pesquisadores geralmente se concentravam num pequeno trecho do genoma do vírus chamado região VP4/2, alinhavam esse trecho com cepas de referência conhecidas, mediam o quão diferentes eram as sequências e então aplicavam valores de corte para decidir a que tipo cada amostra pertencia. Mas essas etapas eram realizadas com uma mistura de programas, edições manuais e julgamento pessoal. Isso dificultava a comparação ou repetição de estudos diferentes, mesmo quando usavam dados semelhantes. O rhinotypeR foi criado especificamente para transformar esse processo em vários passos e sujeito a erros em um fluxo de trabalho único e roteirizado que qualquer pessoa pode executar e compartilhar.
O que o novo software realmente faz
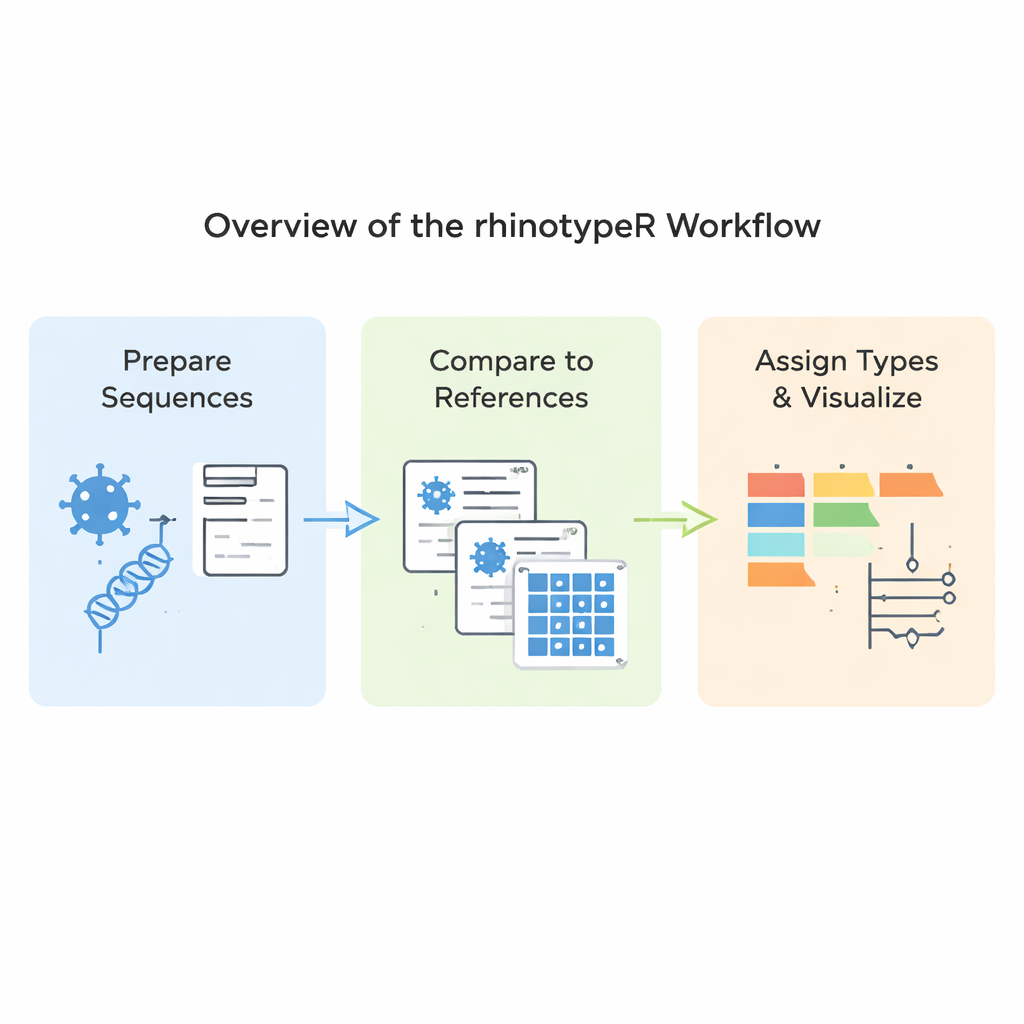
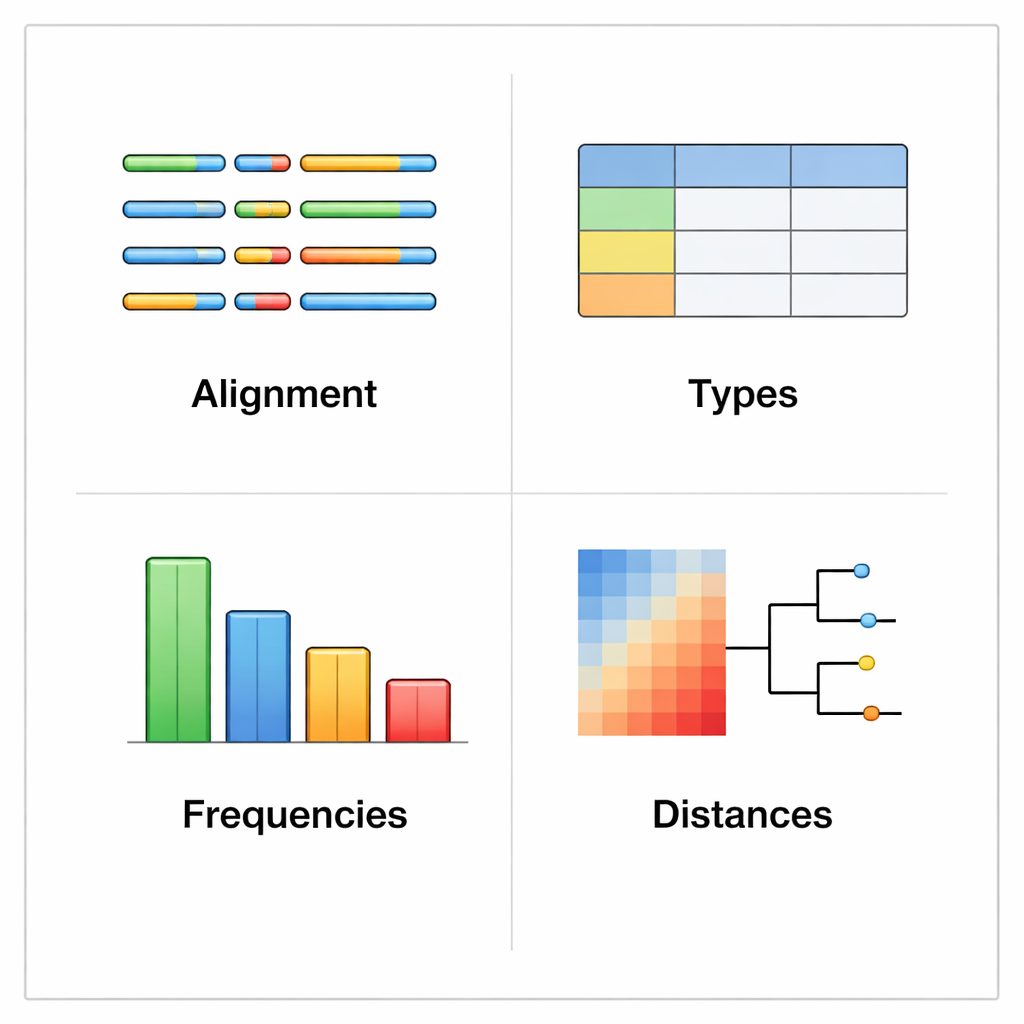
rhinotypeR roda dentro do ambiente amplamente usado R e Bioconductor para análise de dados. Ele recebe um conjunto de sequências VP4/2 de rinovírus e as conduz por três etapas principais: preparar e alinhar as sequências, calcular a distância de cada uma até um conjunto curado de tipos de referência e então atribuir cada amostra ao tipo conhecido mais próximo ou marcá‑la como “não atribuída” se for muito diferente. A mesma ferramenta pode gerar saídas visuais, incluindo mapas codificados por cor das diferenças genéticas, árvores filogenéticas simples e gráficos mostrando a prevalência de cada tipo num conjunto de dados. Usuários podem alinhar seus dados com programas externos se preferirem, ou deixar que o rhinotypeR cuide de todo o processo dentro do R para máxima reprodutibilidade.
Colocando a ferramenta à prova
Para verificar que o rhinotypeR produz resultados confiáveis, os autores compararam suas medidas de distância com as de dois programas estabelecidos, ape e MEGA X, usando os mesmos arquivos de entrada e modelos. Os resultados coincidiram quase perfeitamente; quaisquer pequenas discrepâncias deveram‑se ao arredondamento normal em cálculos de computador, não a diferenças reais de método. A equipe então executou o rhinotypeR em uma grande coleção de mais de 2.300 sequências de rinovírus de estudos anteriores, abrangendo mais de 90% dos tipos conhecidos. Em cerca de quatro em cada cinco casos, a nova ferramenta concordou exatamente com os rótulos de tipo anteriores. A maioria das discordâncias ocorreu justamente em torno dos pontos de corte pré‑acordados usados para separar um tipo do outro, precisamente onde decisões limítrofes são mais esperadas. Importante: amostras que não puderam ser atribuídas com confiança a um tipo conhecido não mostraram sinais de serem simplesmente de baixa qualidade ou conterem pouco vírus, sugerindo que podem refletir uma diversidade viral genuína.
Por que isso importa para a saúde pública
Para não especialistas, a mensagem principal é que o rhinotypeR não reinventa como os cientistas classificam os vírus do resfriado; em vez disso, torna esse processo mais claro, transparente e fácil de reproduzir. Ao agrupar alinhamento, cálculos de distância e atribuição de tipos em um único pacote de código aberto — junto com resumos visuais claros — ele ajuda pesquisadores e programas de vigilância a processar milhares de amostras de forma consistente. Essa consistência melhora nossa capacidade de comparar estudos de lugares e épocas diferentes, detectar cedo linhagens virais incomuns ou emergentes e ligar padrões genéticos a tendências reais de doença. A longo prazo, ferramentas como o rhinotypeR fortalecem o monitoramento rotineiro de resfriados aparentemente comuns que, em muitas pessoas, podem desencadear doenças graves.
Citação: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Palavras-chave: genotipagem de rinovírus, vigilância molecular, sequenciamento VP4/2, ferramentas de bioinformática, vírus respiratórios