Clear Sky Science · pt
Uma abordagem de aprendizado de máquina para prever coeficientes osmóticos e derivar coeficientes de atividade em sais de amônio alquila
Produtos cotidianos com complexidade oculta
De amaciantes e condicionadores de cabelo a lenços desinfetantes e enxaguantes bucais, uma família de substâncias chamada sais de amônio quaternário — frequentemente abreviada para “Quats” — atua discretamente em muitos produtos de que dependemos. Eles ajudam a eliminar germes, amaciar roupas e acelerar reações industriais. Ainda assim, prever exatamente como esses sais se comportam em água tem sido surpreendentemente difícil, limitando a eficiência com que podemos projetar formulações mais seguras e sustentáveis. Este estudo mostra como o aprendizado de máquina moderno pode aprender com medições passadas para prever esse comportamento de forma mais flexível e, em muitos casos, com mais precisão do que modelos tradicionais.
Por que esses sais importam
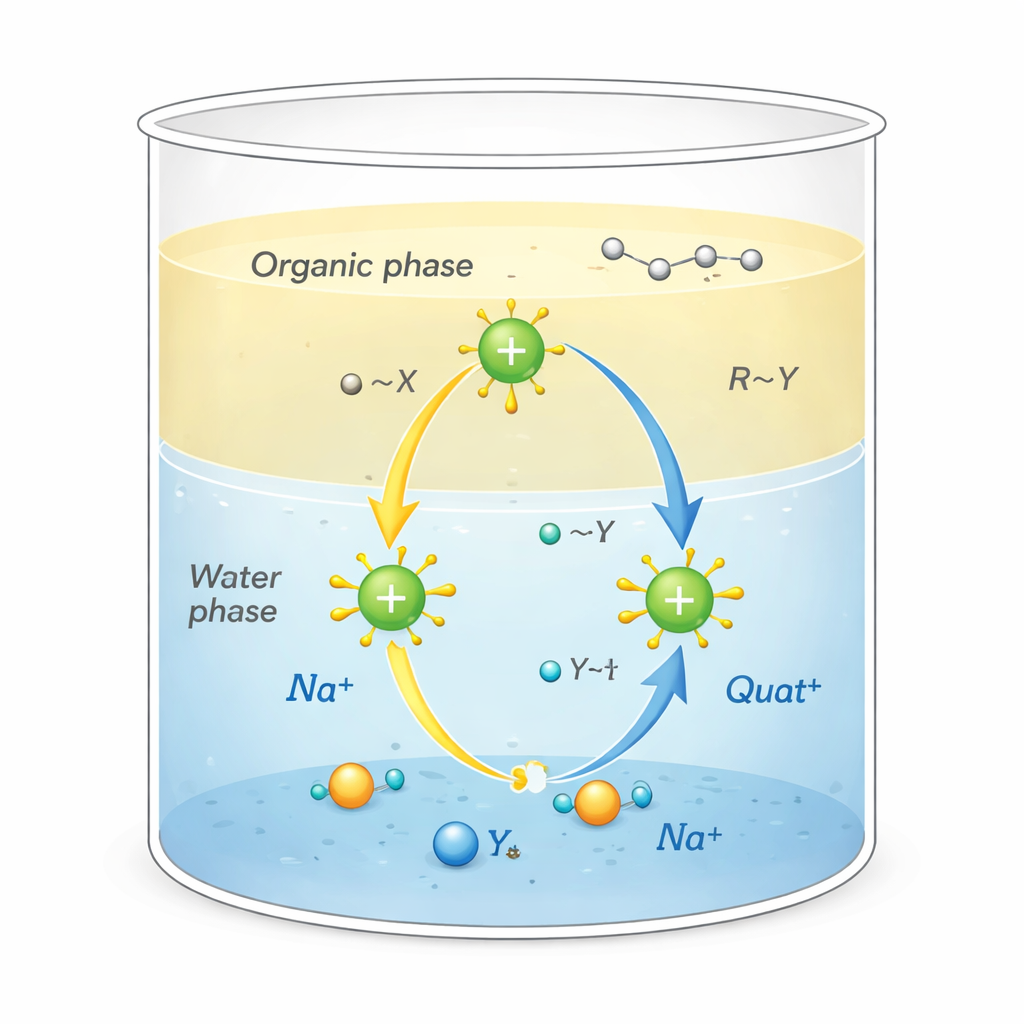
Os Quats são moléculas carregadas positivamente rodeadas por “caudas” ricas em carbono. Essa forma incomum permite que desempenhem várias funções ao mesmo tempo: aderir a sujeira oleosa, grudar em superfícies como tecidos ou cabelos e desorganizar membranas de microrganismos, tornando‑os desinfetantes e surfactantes poderosos. Também são usados como catalisadores de transferência de fase, atuando como veículos que transportam íons reativos da água para solventes mais oleosos onde normalmente não iriam. Essa ação de transporte, que ocorre na fronteira entre água e óleo, pode acelerar dramaticamente reações químicas usadas na fabricação de fármacos, polímeros e produtos químicos finos.
Por que é difícil prever seu comportamento
Para projetar novos Quats ou ajustar os existentes, os químicos precisam saber como eles se comportam em solução — quão fortemente interagem com a água e com outros íons dissolvidos. Duas medidas-chave são o coeficiente osmótico, que reflete como sais afetam a tendência da água de atravessar membranas, e o coeficiente de atividade, que captura quão “efetiva” é uma espécie dissolvida em comparação com uma solução ideal, perfeitamente misturada. Tradicionalmente, esses valores são obtidos por experimentos meticulosos ou usando modelos físicos complexos como Electrolyte‑NRTL e Extended UNIQUAC, que exigem muitos parâmetros ajustados e não são fáceis de generalizar para novas moléculas.
Ensinando um computador a “ler” moléculas
Os pesquisadores seguiram um caminho diferente: perguntaram se um computador poderia aprender a relação entre a estrutura dos Quats e o comportamento osmótico diretamente a partir de dados existentes. Reuniram 1.654 medições de coeficientes osmóticos para 52 Quats diferentes a partir da literatura científica. Cada molécula foi descrita usando a notação SMILES — uma representação em string que codifica características como o número de átomos de carbono e oxigênio, presença de anéis benzênicos, ramificações e o tipo de grupo de nitrogênio carregado positivamente, além do íon negativo acompanhante (como cloreto, brometo ou nitrato). Esses descritores estruturais, mais a concentração do sal, serviram como entradas para diversos algoritmos de aprendizado de máquina supervisionado implementados em Python.
Encontrando o preditor mais confiável
Sete algoritmos diferentes, incluindo regressão linear, árvores de decisão, florestas aleatórias, máquinas de vetor de suporte, gradient boosting, k‑nearest neighbors e processos Gaussianos, foram treinados em 70% dos dados e testados nos 30% restantes. A equipe também empregou um esquema de validação mais rigoroso em que todos os dados de um sal eram excluídos para avaliar o quão bem os modelos extrapolavam para um composto realmente não visto. A regressão linear teve desempenho ruim, não capturando tendências importantes não lineares. Métodos baseados em árvores ajustaram muito bem os dados de treinamento, mas produziram previsões ligeiramente irregulares e perderam precisão em sais novos. O modelo de processo Gaussiano alcançou o melhor equilíbrio: entregou curvas suaves e fisicamente plausíveis para os coeficientes osmóticos e obteve um erro percentual absoluto médio de aproximadamente 5% no geral, superando abordagens alternativas de aprendizado de máquina nos testes mais exigentes.
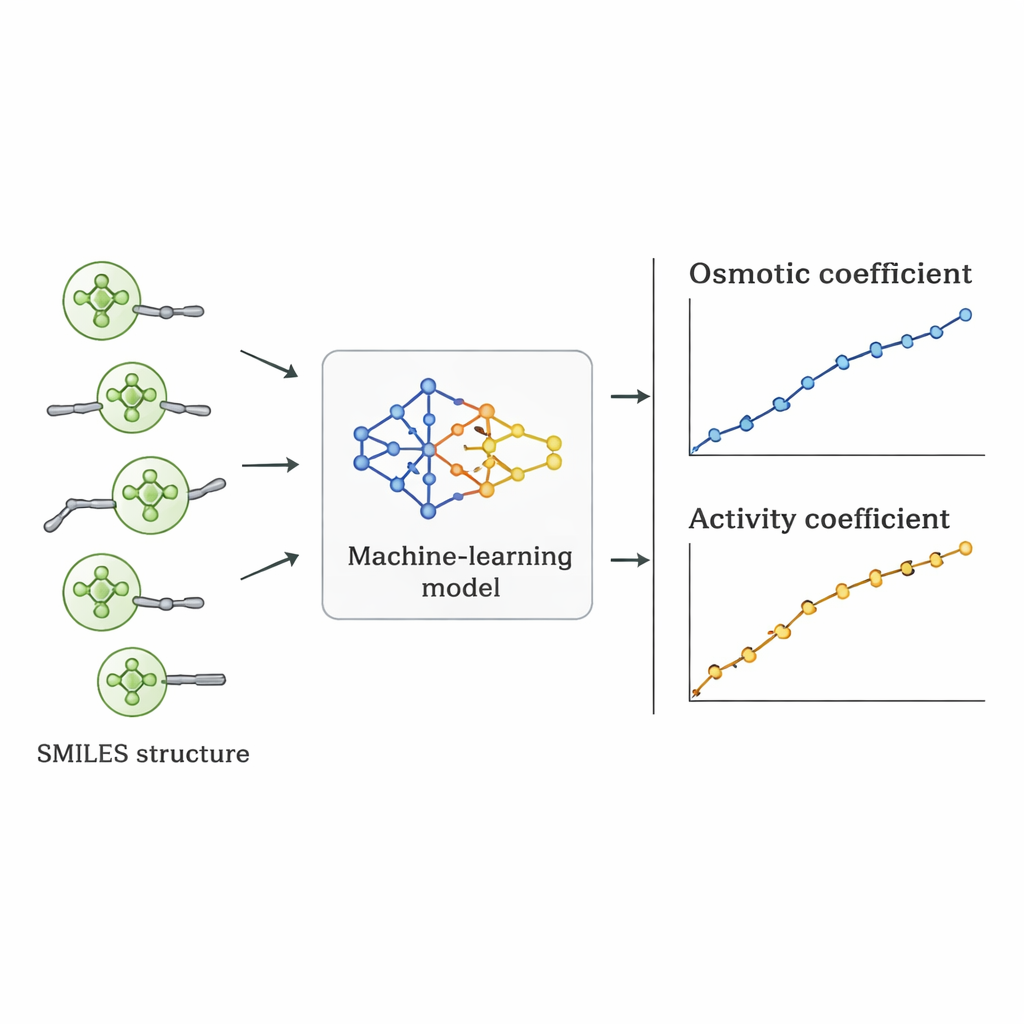
Do comportamento osmótico a números úteis de projeto
Uma vez escolhido o melhor modelo, seus coeficientes osmóticos previstos foram convertidos em coeficientes de atividade usando relações termodinâmicas padrão. Quando esses coeficientes de atividade foram comparados com valores derivados de experimentos e de modelos físicos estabelecidos, a abordagem de aprendizado de máquina muitas vezes os igualou ou superou para Quats individuais. Embora seu erro médio ao longo de todas as substâncias fosse ligeiramente maior do que o de alguns modelos especializados, ela tinha uma vantagem crucial: por ser guiada por descritores estruturais em vez de ajustes específicos por sal, pode ser aplicada a novos Quats nunca medidos em laboratório, desde que suas estruturas se assemelhem às do conjunto de treinamento.
O que isso significa para produtos e processos
Para um público não especializado, a mensagem é que computadores agora podem “ler” descrições compactas em texto de moléculas e, a partir de padrões aprendidos em dados passados, prever como essas moléculas se comportarão em água com precisão impressionante. Isso abre a porta para triagens mais rápidas e baratas de novos Quats para desinfetantes, produtos de limpeza, cuidados pessoais e catalisadores industriais, sem a necessidade de experimentação exaustiva para cada candidato. O modelo atual é apenas um primeiro passo, e os autores observam que impressões moleculares mais ricas e algoritmos mais recentes poderiam melhorar ainda mais o desempenho. Mesmo assim, demonstra como ferramentas orientadas por dados podem complementar a química tradicional, ajudando engenheiros a projetar formulações mais eficazes e potencialmente mais seguras ao explorar possibilidades químicas que seriam impraticáveis de testar uma a uma no laboratório.
Citação: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Palavras-chave: sais de amônio quaternário, catálise por transferência de fase, coeficientes osmóticos, coeficientes de atividade, aprendizado de máquina em química