Clear Sky Science · pt
Estrutura híbrida de deep learning para classificação precisa de dados genômicos de alta dimensionalidade
Entendendo a enxurrada de dados genômicos
As tecnologias modernas de DNA podem medir dezenas de milhares de genes em um único experimento, prometendo detecção precoce de doenças e tratamentos mais precisos. Ainda assim, essa abundância de dados é tão grande, ruidosa e complexa que até modelos computacionais poderosos frequentemente têm dificuldade em encontrar padrões claros e confiáveis. Este artigo apresenta um novo tipo de sistema de inteligência artificial (IA) projetado especificamente para lidar com esses dados genômicos avassaladores, com o objetivo de tornar as previsões mais precisas e, ao mesmo tempo, explicar como essas previsões foram feitas.
Por que os dados genômicos são tão difíceis de usar
Estudos genômicos rotineiramente produzem muito mais medições do que pacientes ou amostras. Muitas dessas medições são irrelevantes, redundantes ou distorcidas por ruído técnico. Métodos tradicionais de aprendizado de máquina ou exigem que especialistas humanos selecionem manualmente quais genes podem importar, ou tentam usar tudo e correm o risco de overfitting — isto é, ter bom desempenho nos dados de treinamento, mas falhar em novos casos. Deep learning, que transformou campos como o reconhecimento de imagens, pode aprender automaticamente padrões a partir de dados brutos. Contudo, em genômica ele frequentemente se comporta como uma caixa preta: pode fornecer respostas precisas, mas oferece pouca visão sobre o porquê, limitando sua aceitação na medicina, onde transparência é essencial.
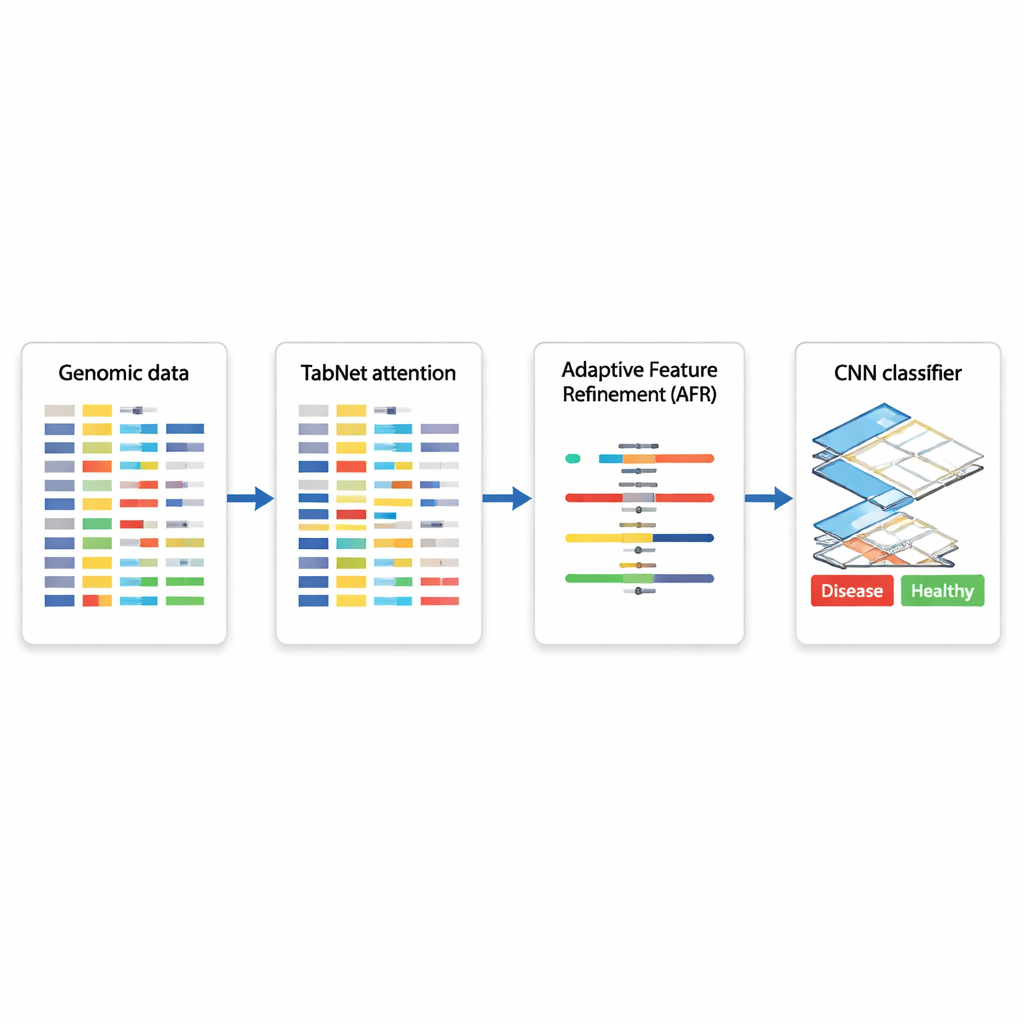
Um projeto híbrido de IA para decisões baseadas em genes
Os autores propõem uma arquitetura híbrida de deep learning que encadeia três módulos especializados. Primeiro, um componente chamado TabNet age como um holofote, vasculhando todas as medições genômicas disponíveis e aprendendo quais recursos são mais informativos para uma dada tarefa — por exemplo, distinguir tecido canceroso de não canceroso. Em vez de tratar cada gene igualmente, o TabNet concentra atenção em um subconjunto esparso que parece mais relevante. Em seguida, uma camada de Refinamento Adaptativo de Recursos (AFR) pega esses sinais selecionados e os repondera, fortalecendo padrões consistentes e significativos enquanto atenua ainda mais o ruído. Finalmente, uma rede neural convolucional (CNN), comumente usada em análise de imagens, examina como os recursos refinados interagem localmente, capturando relações sutis entre grupos de genes que podem sinalizar um determinado subtipo de doença ou estado biológico.
Colocando o modelo à prova
A estrutura foi avaliada em três grandes recursos públicos: um conjunto de dados de câncer de mama do The Cancer Genome Atlas, um conjunto de células únicas de melanoma do Gene Expression Omnibus e um conjunto epigenômico do projeto ENCODE. Juntas, essas coleções incluem milhares de amostras e dezenas de milhares de características por amostra, cobrindo atividade gênica e marcas químicas no DNA. Em todos os conjuntos de dados, o modelo híbrido superou várias abordagens de ponta, melhorando a acurácia e medidas-chave de qualidade de classificação, como a área sob a curva ROC (AUC) e a pontuação F1 em cerca de 5 a 8 pontos percentuais. Importante, esses ganhos não vieram ao custo de transparência: o modelo produz mapas de atenção do TabNet e mapas de ativação da CNN que destacam quais genes e regiões foram mais influentes em cada predição.
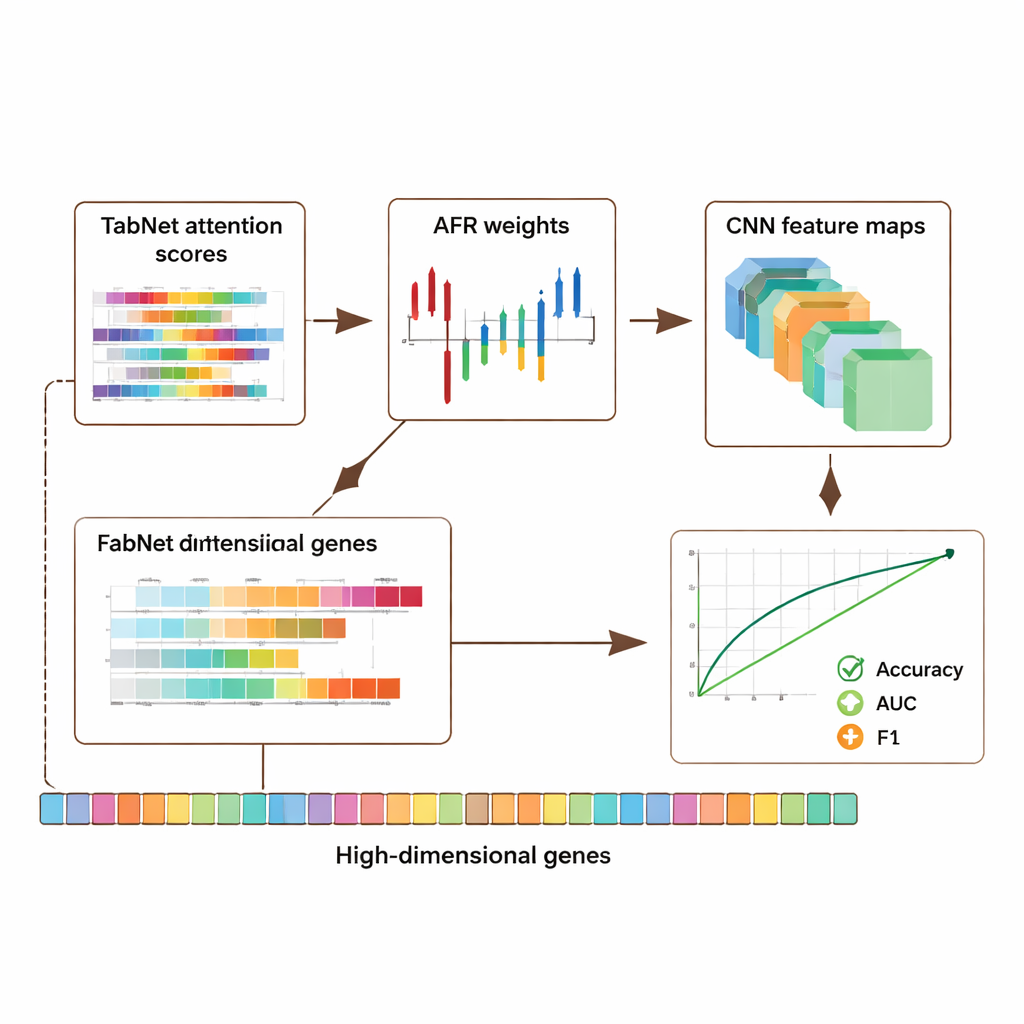
Equilibrando precisão, privacidade e confiança
Como dados genômicos são profundamente pessoais, os autores também investigaram como proteger a privacidade mantendo o sinal útil. Eles introduziram um mecanismo de privacidade adaptativo que adiciona mais ruído a características altamente sensíveis e menos a outras, combinado com mascaramento de entradas selecionadas. Testes mostraram que mesmo quando ruído moderado foi introduzido, o modelo manteve forte acurácia e discriminação, com desempenho degradando-se de forma gradual à medida que a proteção aumentava. Ao mesmo tempo, os padrões interpretáveis de atenção e ativação frequentemente apontaram para genes já conhecidos por desempenhar papéis no câncer e na regulação imune, sugerindo que o sistema não está apenas memorizando dados, mas capturando sinais biologicamente significativos. Um estudo de ablação — removendo sistematicamente partes da arquitetura — confirmou que cada módulo, especialmente a camada AFR, deu uma contribuição mensurável ao desempenho.
O que isso significa para a medicina do futuro
Em termos simples, este trabalho oferece uma maneira mais inteligente de vasculhar enormes planilhas genômicas para encontrar padrões ligados a doenças, ao mesmo tempo em que mostra quais entradas na planilha foram mais importantes. Ao combinar seleção direcionada de recursos, refinamento cuidadoso e reconhecimento de padrões, o modelo híbrido melhora a precisão das previsões, permanece gerenciável computacionalmente e fornece pistas visuais que clínicos e biólogos podem interpretar. Embora sejam necessários mais testes em grupos de pacientes mais amplos e diversos, tais estruturas poderiam ajudar a identificar novos biomarcadores, refinar subtipos de doença e apoiar ferramentas de decisão clínica em medicina de precisão — aproximando a análise de DNA por IA de um uso no mundo real.
Citação: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Palavras-chave: deep learning genômico, descoberta de biomarcadores do câncer, IA interpretável, medicina de precisão, genômica preservadora de privacidade