Clear Sky Science · pt
Um estudo quantitativo de compostos citotóxicos usando descritores baseados em grafos e aprendizado de máquina
Por que esta pesquisa importa para os futuros medicamentos contra o câncer
Medicamentos contra o câncer que matam células tumorais, conhecidos como fármacos citotóxicos, frequentemente caminham na corda bamba entre salvar vidas e causar efeitos colaterais graves. Para projetar tratamentos mais seguros e eficazes, os cientistas precisam de maneiras rápidas e confiáveis de prever como esses fármacos se movem pelo corpo — quão bem são absorvidos, com que facilidade atravessam membranas celulares e onde acabam se acumulando. Este estudo mostra como descrições matemáticas de moléculas farmacêuticas, combinadas com aprendizado de máquina moderno, podem estimar com precisão uma propriedade-chave que controla esse comportamento, potencialmente acelerando a busca por terapias anticâncer melhores.
Uma superfície-chave que controla aonde os fármacos podem ir
Uma ideia central do artigo é a área polar de superfície topológica, ou Top_PSA. Em termos simples, este é um número que reflete quanto da superfície de uma molécula é composto por regiões “polares” — partes que gostam de água e podem formar ligações de hidrogênio. Moléculas com áreas polares de superfície muito altas frequentemente têm dificuldade para atravessar membranas lipídicas e podem ser mal absorvidas quando administradas por via oral. Moléculas com áreas polares de superfície muito baixas podem atravessar muitas barreiras com facilidade excessiva, às vezes causando efeitos indesejados em tecidos sensíveis, como o cérebro. Top_PSA tornou-se um atalho popular para estimar essas propriedades de transporte porque pode ser calculada rapidamente a partir de um desenho 2D da molécula, sem a necessidade de simulações 3D lentas.
Transformando desenhos moleculares em números
Os pesquisadores reuniram um conjunto curado de 156 compostos citotóxicos diferentes, extraídos de fármacos anticâncer reais e de agentes experimentais. Em seguida, converteram cada molécula em 58 chamados descritores — números que capturam características como quantos átomos ela possui, quantos anéis, quão flexíveis são suas ligações, quantos átomos podem formar ligações de hidrogênio e quão polares ou eletronegativos são diferentes segmentos. Muitos desses descritores vêm da teoria dos grafos, onde uma molécula é tratada como uma rede de nós e ligações conectados. Esse retrato numérico rico de cada molécula serviu como entrada para modelos computacionais que tentaram prever os valores de Top_PSA calculados por ferramentas químicas amplamente usadas.
Testando múltiplos caminhos para uma previsão precisa
Para encontrar a melhor forma de vincular esses descritores ao Top_PSA, a equipe comparou várias estratégias de modelagem. Eles testaram regressão linear padrão, bem como duas versões “regularizadas” chamadas regressão ridge e LASSO, projetadas para lidar melhor com informações ruidosas e sobrepostas. Também exploraram diferentes esquemas de preparação de dados: ajustar modelos diretamente aos descritores brutos, comprimí‑los com análise de componentes principais (PCA), escalá‑los de forma robusta para reduzir o impacto de valores extremos (robust scaling), ajustar outliers e podar características altamente correlacionadas usando uma medida chamada fator de inflação de variância. Cada abordagem foi avaliada cuidadosamente usando validação cruzada k‑fold, um método que divide repetidamente os dados em subconjuntos de treinamento e teste para proteger contra overfitting.
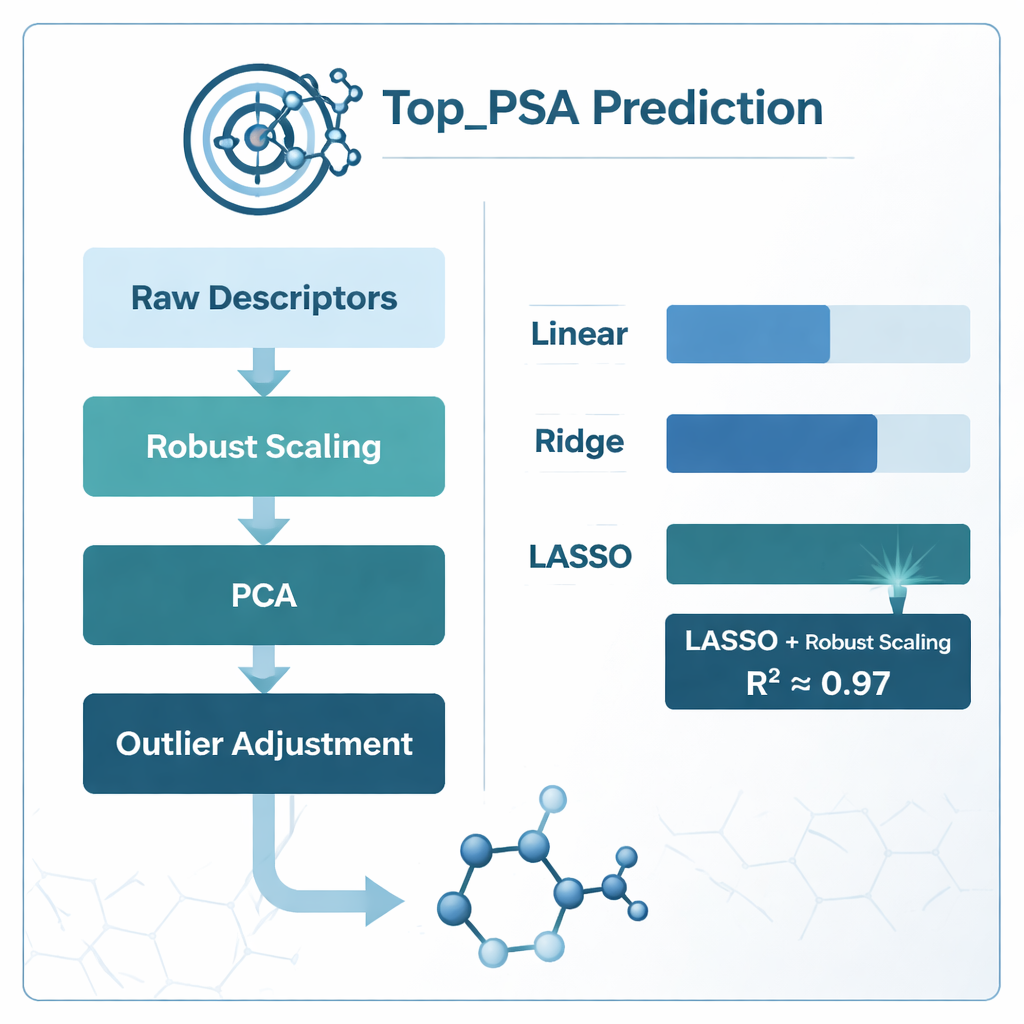
O que funcionou melhor e o que os modelos aprenderam
O vencedor claro foi a combinação de escalamento robusto com regressão LASSO, que alcançou um coeficiente de determinação (R²) de cerca de 0,97 — o que significa que pôde explicar aproximadamente 97% da variação em Top_PSA entre os 156 fármacos. Modelos baseados em PCA chegaram perto na precisão bruta, mas eram mais difíceis de interpretar quimicamente porque os descritores originais ficam misturados em componentes abstratos. A simples poda de descritores correlacionados usando o fator de inflação de variância na verdade prejudicou o desempenho, sugerindo que algumas medidas sobrepostas ainda carregam informação química útil. Ao examinar quais pesos de descritor o LASSO manteve não nulos, os autores descobriram que os fatores mais importantes eram a presença de heteroátomos como nitrogênio e oxigênio, a capacidade de doar ou aceitar ligações de hidrogênio e índices que acompanham como átomos eletronegativos estão distribuídos ao longo do grafo molecular — todas características que correspondem ao entendimento químico intuitivo da área polar de superfície.
Como isso pode orientar um melhor desenho de fármacos
Para leitores fora da área, a mensagem chave é que impressões digitais matemáticas bem preparadas de moléculas, quando pareadas com métodos de aprendizado de máquina bem escolhidos, podem fornecer estimativas rápidas e confiáveis de quão “aderentes” ou “deslizantes” serão os fármacos anticâncer enquanto viajam pelo corpo. O estudo oferece orientações práticas para outros pesquisadores sobre como pré-processar dados de descritores, quais abordagens de modelagem favorecer e quais atalhos evitar. A longo prazo, modelos de Top_PSA robustos e interpretáveis podem ajudar químicos a filtrar enormes bibliotecas virtuais de potenciais fármacos, concentrando seus esforços em compostos com o equilíbrio certo entre atravessamento de membrana e segurança — um passo importante rumo a tratamentos contra o câncer mais eficazes e menos tóxicos.
Citação: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Palavras-chave: fármacos citotóxicos, área polar de superfície, descritores moleculares, aprendizado de máquina, permeabilidade de fármacos