Clear Sky Science · pt
Disfunção mitocondrial e desregulação de Ca2+ em neurônios humanos derivados de iPSC com mutação em presenilina-1 surgem sob estresse por um mecanismo independente de MCU-1
Por que isso importa para a doença de Alzheimer
A doença de Alzheimer costuma ser descrita em termos de placas proteicas pegajosas no cérebro, mas muito antes da memória declinar, as pequenas “usinas” dentro dos neurônios — as mitocôndrias — e o manejo dos íons de cálcio podem já estar falhando. Este estudo usa neurônios humanos cultivados a partir de células da pele de uma pessoa portadora de uma conhecida mutação familiar de Alzheimer para fazer uma pergunta simples, porém crucial: quão cedo, e de que maneira, a produção de energia e o equilíbrio do cálcio começam a falhar?
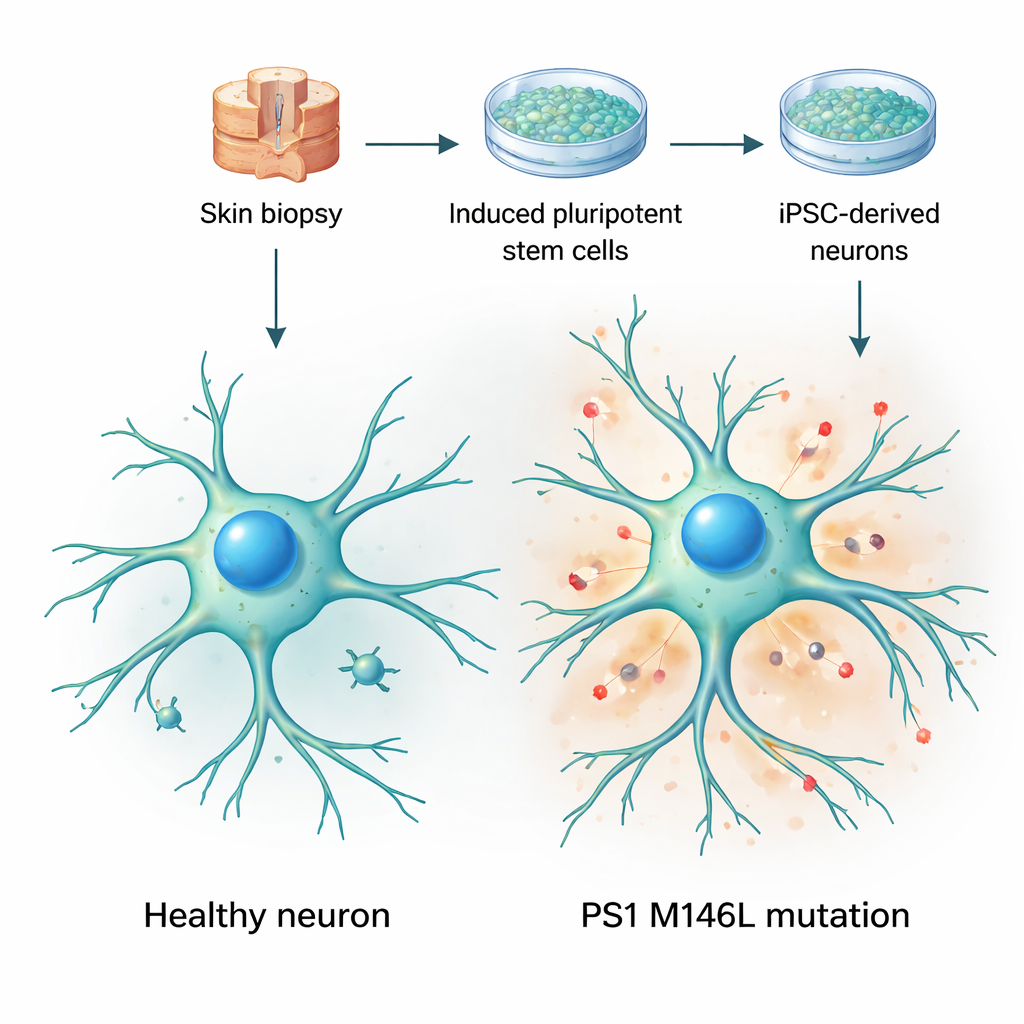
Transformando células da pele em modelos cerebrais vivos
Os pesquisadores começaram com biópsias de pele de duas mulheres: uma voluntária saudável e uma portadora assintomática de uma mutação em presenilina‑1 chamada M146L, presente em uma família argentina com Alzheimer de início precoce. Eles reprogramaram as células da pele em células-tronco pluripotentes induzidas — células que podem se tornar quase qualquer tecido — e então as induziram a se diferenciar em neurônios. Ao longo de várias semanas em cultura, essas células adquiriram formas neurais típicas, estenderam longos processos ramificados e expressaram marcadores neuronais padrão. Importante, tanto as células controle quanto as mutantes amadureceram em taxas semelhantes e pareciam, de modo geral, saudáveis, o que permitiu à equipe focar em mudanças funcionais sutis em vez de perda ou dano celular óbvios.
Sinais elétricos e cálcio sob tensão
Os neurônios dependem do controle rigoroso do cálcio, um átomo carregado que atua como um interruptor rápido para muitos processos celulares. Usando corantes fluorescentes, a equipe acompanhou como os níveis de cálcio mudavam dentro das células quando estas eram estimuladas eletricamente com potássio ou ativadas com moléculas sinalizadoras. Sob estimulação despolarizante simples, os neurônios portadores da mutação M146L mostraram aumentos de cálcio mais fracos do que os neurônios controle, sugerindo problemas em manter os gradientes elétricos e iônicos que normalmente impulsionam a entrada de cálcio. Entretanto, quando os pesquisadores desencadearam uma situação mais estressante — forçando o vazamento de cálcio de reservatórios internos do retículo endoplasmático — a diferença ficou mais clara. Em resposta a esse estresse, as mitocôndrias dos neurônios mutantes captaram visivelmente menos cálcio do que as das células controle, indicando uma capacidade reduzida de amortecer surtos perigosos de cálcio.
Desacoplando o uso de energia do equilíbrio do cálcio
Para entender como esse manejo alterado do cálcio afeta o metabolismo celular, os investigadores mediram quanto oxigênio os neurônios consumiam — um proxy direto da atividade mitocondrial. Surpreendentemente, os neurônios com a mutação M146L consumiam mais oxigênio: suas taxas basais e máximas de consumo de oxigênio, e a quantidade de oxigênio vinculada à produção de ATP, foram todas maiores do que nas células controle. Ainda assim, a eficiência no acoplamento entre o uso de oxigênio e a produção de ATP parecia semelhante, e não houve aumento no número de mitocôndrias ou nas principais enzimas produtoras de ATP. Em vez disso, as mitocôndrias nos neurônios mutantes eram mais longas e tubulares, com níveis mais altos de uma proteína de fusão chamada mitofusina‑1, um padrão frequentemente observado em células sob estresse crônico de baixo grau. Essas mitocôndrias hiperativas e alongadas também geraram mais espécies reativas de oxigênio, moléculas instáveis que podem danificar proteínas e DNA se não forem adequadamente controladas.
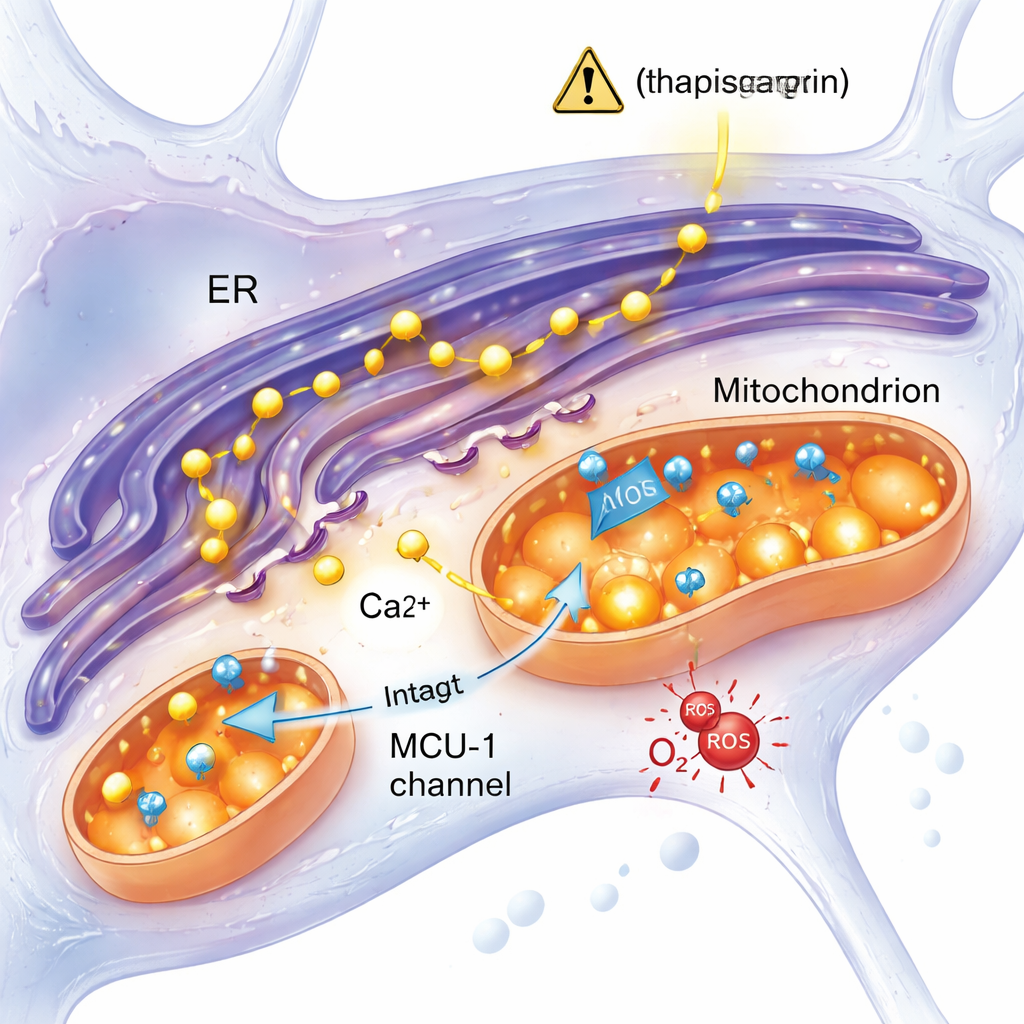
Uma resposta ao estresse independente de um canal-chave de cálcio
Uma ideia dominante na pesquisa sobre Alzheimer é que o excesso de cálcio proveniente do retículo endoplasmático corre para as mitocôndrias através de um canal chamado uniportador mitocondrial de cálcio (MCU‑1), sobrecarregando‑as e levando à disfunção. Este estudo testou essa noção diretamente. Quando a equipe bloqueou o MCU‑1 com um inibidor específico, tanto neurônios controle quanto mutantes mostraram forte redução na captação mitocondrial de cálcio, confirmando que o canal em si funcionava em ambos os grupos. Além disso, quando a liberação de cálcio foi desencadeada por uma via mais fisiológica envolvendo o receptor IP3 — outro portão importante de cálcio — as células mutantes e controle responderam de forma semelhante. Esses resultados afastam a hipótese de um MCU‑1 defeituoso e, em vez disso, sugerem que os contatos físicos e funcionais entre o retículo endoplasmático e as mitocôndrias, ou outros aspectos de sua interação, estão alterados nos neurônios mutantes.
O que isso significa para entender e tratar a doença
Em conjunto, os achados pintam o quadro de neurônios humanos portadores da mutação PS1 M146L de Alzheimer como células que parecem normais em repouso, mas reagem de maneira anômala sob estresse. Suas mitocôndrias deixam de captar cálcio suficiente quando reservatórios internos são liberados subitamente, ainda que funcionem em ritmo mais acelerado — consumindo mais oxigênio e gerando mais espécies reativas de oxigênio — como se estivessem presas a um modo compensatório custoso. Como isso ocorre em neurônios derivados de humanos vivos antes de quaisquer sintomas clínicos, o trabalho apoia a ideia de que a sinalização de cálcio interrompida e o excesso precoce de trabalho mitocondrial são eventos iniciais na doença de Alzheimer, e não apenas subprodutos tardios. Para o público não especializado, a mensagem-chave é que manter o equilíbrio entre os sinais de cálcio e a produção de energia mitocondrial pode ser tão central para prevenir a doença quanto mirar as placas amilóides mais conhecidas.
Citação: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Palavras-chave: Doença de Alzheimer, mitocôndrias, sinalização de cálcio, mutação em presenilina-1, neurônios derivados de iPSC