Clear Sky Science · pt
Desregulação da homeostase do ferro intracelular por disfunção mitocondrial associada à supressão da expressão de ATP13A2
Por que o ferro dentro das células cerebrais importa
A doença de Parkinson é mais conhecida por tremores e rigidez nos movimentos, mas lá no interior das células cerebrais afetadas ocorre outro drama: o ferro, um metal essencial, começa a se acumular onde não deveria. Este estudo faz uma pergunta simples, porém importante: como esse acúmulo de ferro acontece e de que forma ele pode danificar as pequenas usinas de energia e os centros de reciclagem dentro dos neurônios? Ao responder, o trabalho oferece pistas sobre por que certas regiões do cérebro degeneram na doença de Parkinson e em distúrbios relacionados, e aponta para novos tipos de tratamentos que vão além de repor a dopamina.
Um olhar mais atento a uma pista genética rara
Os pesquisadores focam numa forma hereditária rara de doença de Parkinson, chamada PARK9, causada por defeitos em um gene denominado ATP13A2. Esse gene codifica uma proteína localizada nos lisossomos, os compartimentos celulares responsáveis pelo processamento de resíduos e reciclagem. Pessoas com mutações em ATP13A2 também podem desenvolver uma condição marcada por depósitos de ferro no cérebro. Essa conexão fez do ATP13A2 um ponto de entrada ideal para estudar como o equilíbrio do ferro se desregula. Usando uma linhagem celular humana semelhante a neurônios que superproduz a proteína associada ao Parkinson, alfa‑sinucleína, a equipe utilizou pequenos fragmentos de RNA para reduzir a expressão de ATP13A2 e então acompanhou como ferro, produção de energia e a saúde celular mudavam.
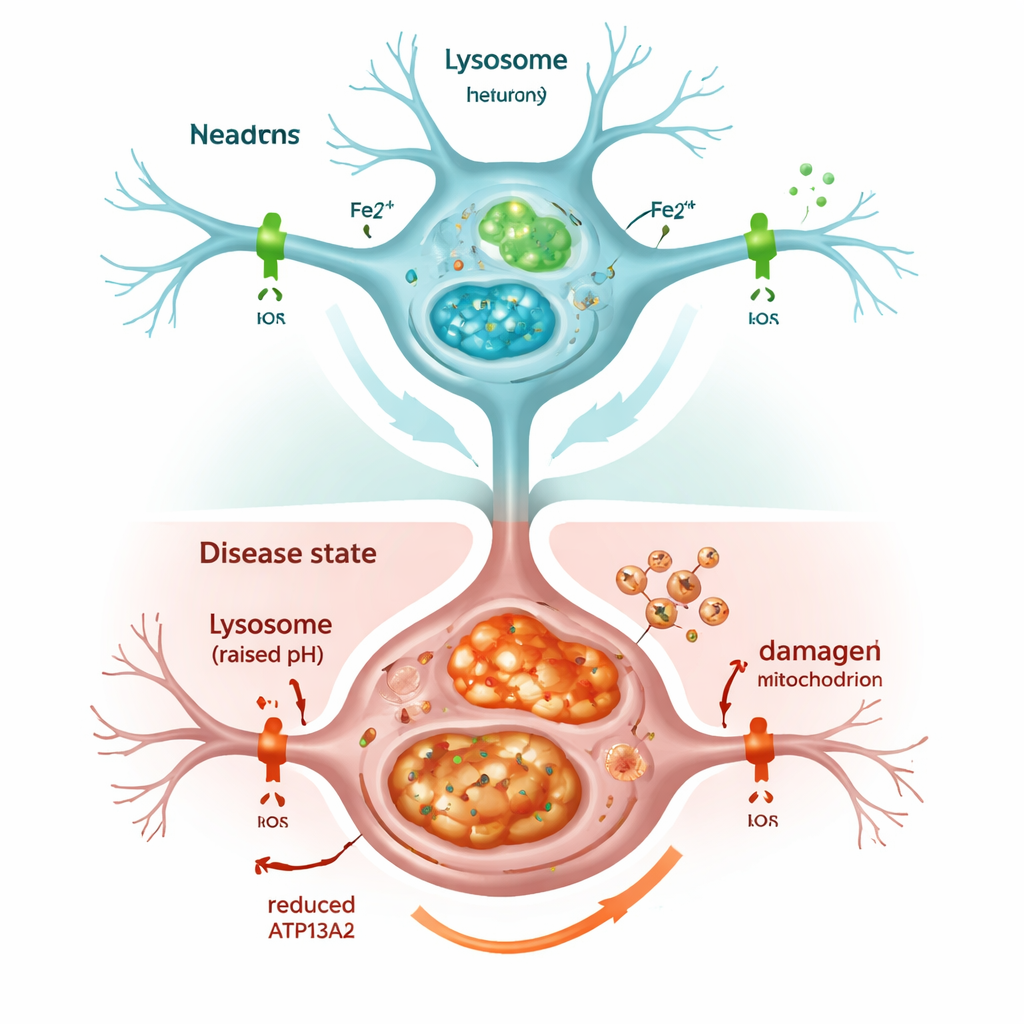
Quando o sistema de reciclagem da célula trava
Silenciar o ATP13A2 enfraqueceu rapidamente os lisossomos. Sua acidez interna, crítica para degradar material indesejado, caiu, e marcadores do processo de limpeza celular, chamado autofagia, se acumularam em vez de serem eliminados. Como resultado, a alfa‑sinucleína se acumulou, refletindo o que é visto nos cérebros de pacientes com Parkinson. As células também mostraram mais ferro no conjunto, e especialmente mais da forma quimicamente ativa, chamada Fe2+, tanto dentro dos lisossomos quanto nas mitocôndrias. A célula respondeu produzindo mais ferritina, uma proteína que armazena ferro, mas isso não foi suficiente para evitar problemas: as mitocôndrias sobrecarregadas geraram excesso de moléculas reativas de oxigênio, e a sobrevivência celular diminuiu. Tratar as células com um composto quelante de ferro, semelhante a alguns usados clinicamente, reduziu esse estresse oxidativo e salvou parcialmente a viabilidade celular, destacando que o excesso de ferro em si era um motor central do dano.
Os sensores de ferro param de ouvir o metal
Normalmente, as células dispõem de um sistema de retroalimentação que detecta quando os níveis de ferro sobem e reage reduzindo a importação de ferro. Uma proteína chamada IRP2 sente o ferro, em parte por meio de um sinal dependente de heme vindo das mitocôndrias, e então ajusta a produção de proteínas responsáveis pelo transporte de ferro na superfície celular. Nas células deficientes em ATP13A2, essa salvaguarda falhou. Os transportadores que trazem ferro para dentro da célula permaneceram altos mesmo com o ferro já elevado. Os níveis da proteína IRP2 quase não mudaram, e adicionar ferro externo não desencadeou sua degradação normal. A equipe rastreou essa falha até as mitocôndrias: mitocôndrias danificadas respiravam com menos eficiência, apresentavam sinais de controle de qualidade defeituoso (mitofagia) e, crucialmente, perderam a capacidade de produzir heme, a molécula contendo ferro que ajuda a IRP2 a detectar o ferro. Sem heme suficiente, a IRP2 não recebeu a mensagem de “ferro em excesso” e permitiu a entrada contínua de ferro.
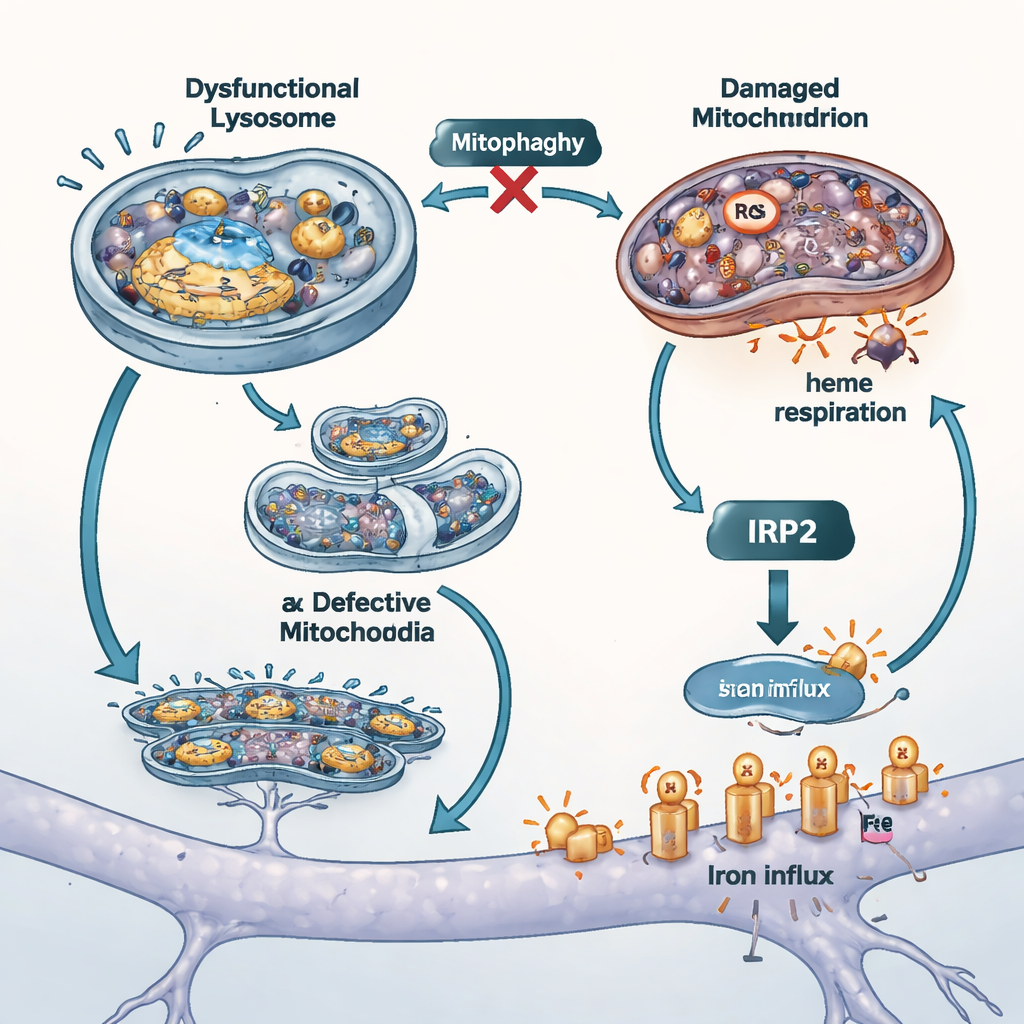
Fechando a torneira do ferro e testando outros modelos
Para sondar quanto essa entrada descontrolada de ferro contribuiu para o dano celular, os cientistas bloquearam duas vias principais de captação de ferro. Usaram uma versão sem ferro da proteína sanguínea transferrina para competir por um importador, e um pequeno fármaco para reduzir a atividade de outro transportador chamado DMT1. Ambas as manobras reduziram o ferro total e o ferro livre dentro das células, diminuíram o estresse oxidativo mitocondrial e melhoraram a sobrevivência, sugerindo que canais de ferro da superfície são amplificadores importantes do dano quando ATP13A2 é perdido. Os pesquisadores também repetiram experimentos-chave em células sem outro gene ligado ao Parkinson, PINK1, conhecido por prejudicar a mitofagia. Essas células exibiram a mesma combinação de acúmulo de ferro e produção de heme enfraquecida, reforçando a ideia de que o controle de qualidade mitocondrial e o equilíbrio do ferro estão intimamente interligados em diferentes formas da doença.
O que isso significa para Parkinson e tratamentos futuros
Em termos simples, o estudo descreve um ciclo vicioso. Quando o ATP13A2 é suprimido, os lisossomos deixam de eliminar componentes danificados, incluindo mitocôndrias defeituosas. Essas mitocôndrias enfraquecidas passam a produzir menos energia e menos heme, prejudicando o sistema celular de detecção de ferro. O ferro continua a entrar pelos transportadores de superfície, se acumula em compartimentos vulneráveis e alimenta reações tóxicas que lesionam ainda mais as mitocôndrias. Ao longo do tempo, esse ciclo pode ajudar a explicar por que certos neurônios morrem na doença de Parkinson e em distúrbios cerebrais com sobrecarga de ferro. As descobertas sugerem que terapias futuras podem não apenas tentar remover o ferro em excesso, mas também restaurar a função adequada dos lisossomos, o controle de qualidade mitocondrial e a produção de heme — atacando o problema em sua origem em vez de apenas limpar o metal depois do dano.
Citação: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Palavras-chave: Doença de Parkinson, ferro cerebral, mitocôndrias, lisossomos, síntese de heme