Clear Sky Science · pt
LASSO Estocástico para dados genômicos de dimensionalidade extremamente alta
Encontrando a agulha no palheiro genômico
A biologia moderna consegue medir dezenas de milhares de genes ao mesmo tempo, mas estudos com pacientes frequentemente incluem apenas algumas centenas de indivíduos. Nessa desigualdade estão escondidos pequenos conjuntos de genes que realmente importam para prever risco de doença ou sobrevida. Este artigo apresenta o “LASSO Estocástico”, um método estatístico projetado para descobrir com confiabilidade esses genes-chave em oceanos de dados genômicos ruidosos, mesmo quando há muito mais genes do que pacientes.
Por que escolher os genes certos é tão difícil
Pesquisadores frequentemente dependem de ferramentas como o LASSO, que encolhem os efeitos de genes irrelevantes em direção a zero ao mesmo tempo em que preservam os mais informativos. Versões clássicas do LASSO, porém, têm dificuldade quando o número de genes supera amplamente o número de amostras, como é comum em genômica do câncer. O LASSO padrão só pode selecionar no máximo tantos genes quanto há pacientes e tende a negligenciar genes que se comportam de forma semelhante entre si. Melhorias anteriores que adicionam penalizações extras conseguem lidar com parte dessa correlação, mas podem também obscurecer o significado biológico ao forçar genes relacionados a agir como se todos influenciassem o resultado na mesma direção.
Construindo amostras aleatórias mais limpas
Uma solução promissora é ajustar o LASSO repetidamente em muitos subconjuntos menores de genes sorteados aleatoriamente e depois combinar os resultados. Ainda assim, essas abordagens de “bootstrap” sofrem de três problemas: genes correlacionados podem se cancelar, muitos genes são raramente ou nunca amostrados, e a aleatoriedade pura torna a seleção final instável. O LASSO Estocástico enfrenta essas questões diretamente com um novo esquema de amostragem chamado bootstrap baseado em correlação. Em vez de escolher genes ao acaso, ele favorece deliberadamente genes que são menos correlacionados com os já escolhidos, gerando conjuntos menores de genes muito mais independentes. Também garante que cada gene seja usado o mesmo número de vezes ao longo das execuções do bootstrap, para que nenhum gene seja injustamente ignorado. 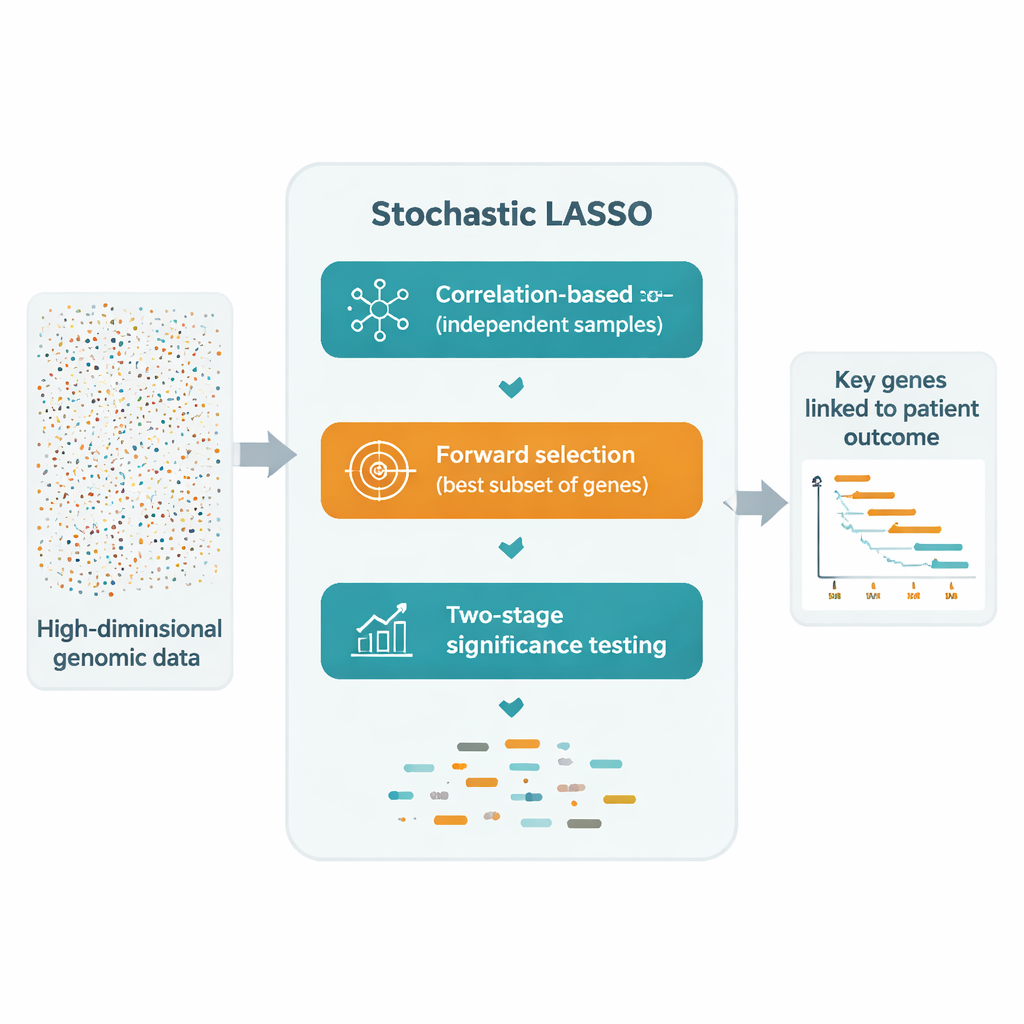
De pistas locais para um conjunto genômico global
Depois de construir esses subconjuntos mais limpos, o LASSO Estocástico registra o tamanho do coeficiente de cada gene ao longo de todas as ajustes do bootstrap. Esse efeito absoluto médio torna-se uma “pontuação local” que reflete quão consistentemente importante o gene aparece. Em vez de testar exaustivamente todas as combinações possíveis, o método constrói modelos candidatos adicionando genes em ordem de suas pontuações locais e avalia o quão bem cada candidato prevê os resultados em dados de validação separados. Dessa forma, ele se estabelece em um conjunto compacto de genes cujos sinais combinados explicam melhor os dados, usando muito menos tentativas do que os métodos tradicionais passo a passo.
Testando quais genes realmente importam
Para passar de “frequentemente selecionado” para “estatisticamente convincente”, os autores introduzem um teste t em duas etapas. Primeiro, checam se o coeficiente médio de cada gene ao longo dos bootstraps é claramente diferente de zero, marcando-o como potencialmente significativo. Em seguida, entre esses candidatos, perguntam se o efeito de cada gene é maior que o tamanho de efeito típico de todos os candidatos. Apenas genes que passam pelos dois testes são declarados significativos. Como esses testes dependem das muitas estimativas do bootstrap, o LASSO Estocástico pode identificar com confiança mais genes significativos do que há pacientes — algo que o LASSO convencional não consegue. 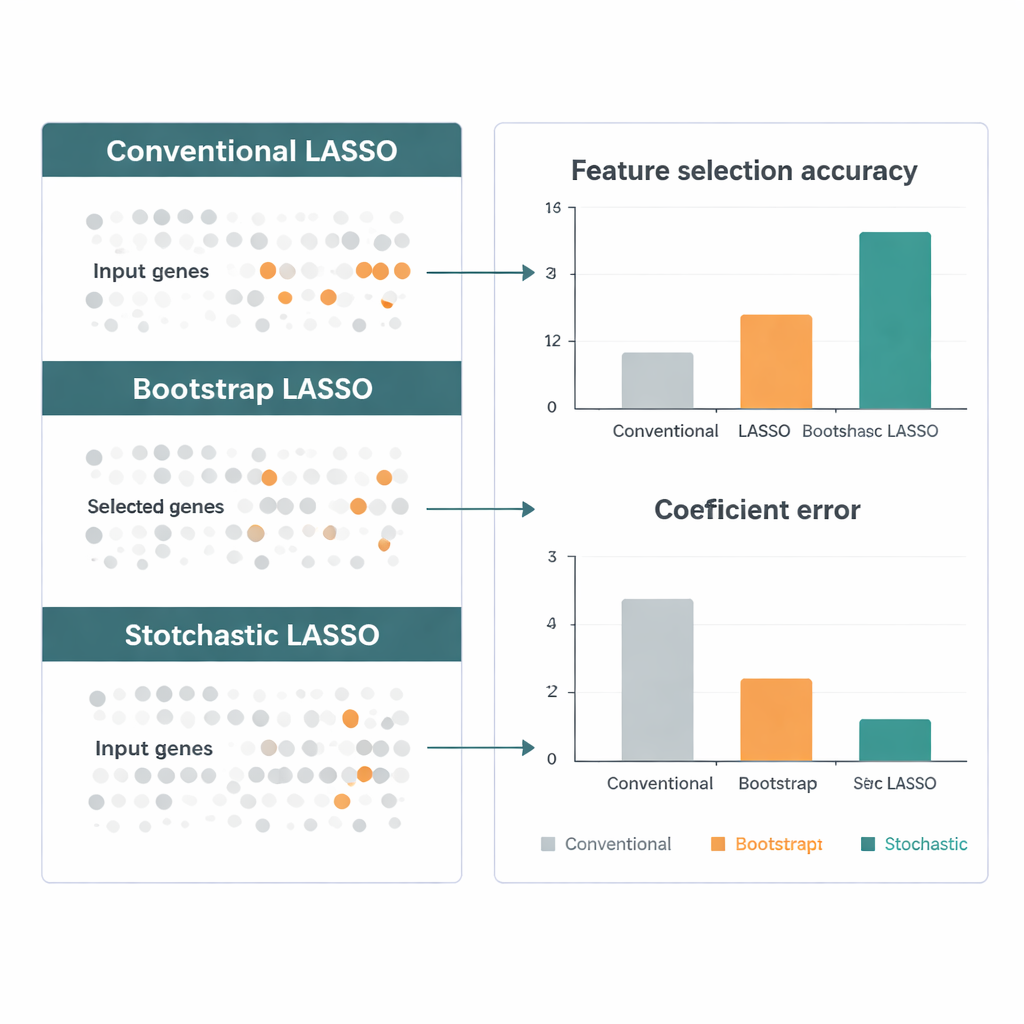
Provando seu valor em simulações e dados de câncer
Os autores comparam o LASSO Estocástico com várias variantes líderes do LASSO usando dados simulados projetados para imitar estudos genômicos reais: muitos genes, fortes correlações e sinais “verdadeiros” conhecidos. Em múltiplos cenários, o novo método encontra os genes corretos com mais frequência, estima seus efeitos com maior precisão e mantém estabilidade entre execuções. Depois, eles aplicam o método a dados de expressão gênica do The Cancer Genome Atlas para tumores cerebrais, incluindo o agressivo glioblastoma. O LASSO Estocástico destaca centenas de genes cuja atividade se relaciona à sobrevida dos pacientes e aponta vias biológicas — como sinalização e rotas de metabolismo de fármacos — que têm suporte independente na literatura, sugerindo que o método não é apenas estatisticamente mais apurado, mas também biologicamente plausível.
O que isso significa para pacientes e pesquisadores
Para não especialistas, a mensagem principal é que o LASSO Estocástico é um filtro mais inteligente para dados genômicos em grande escala. Ele ajuda cientistas a separar genes realmente relacionados à doença do ruído estatístico, mesmo quando os dados são limitados e os genes estão altamente interconectados. Ao fornecer listas de genes e estimativas de efeito mais precisas e mais estáveis, ele pode afiar a busca por biomarcadores, alvos de drogas e assinaturas prognósticas em câncer e outras doenças complexas. Embora demonstrado em regressão linear, a mesma estrutura pode ser integrada a modelos de sobrevivência e problemas de classificação, ampliando seu potencial impacto na pesquisa biomédica.
Citação: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Palavras-chave: seleção de características genômicas, dados de alta dimensionalidade, métodos LASSO, expressão gênica do câncer, descoberta de biomarcadores