Clear Sky Science · pt
O lactato regula o eixo YTHDF2-FTH1 para promover a ferroptose de cardiomiócitos e agravar a lesão por isquemia-reperfusão miocárdica
Por que pacientes cardíacos devem se importar com essa química
Quando médicos reabrem uma artéria coronária bloqueada após um ataque cardíaco, o fluxo súbito de sangue salva músculo, mas também pode causar dano adicional, conhecido como lesão por isquemia–reperfusão. Este estudo revela um culpado surpreendente dentro das células cardíacas: o comum subproduto metabólico lactato. Os autores mostram que o lactato pode ativar um interruptor molecular que empurra as células do coração para um tipo específico de morte celular dirigida pelo ferro, agravando a lesão. Entender essa via oculta pode apontar para novos fármacos que protejam melhor o coração durante o tratamento de emergência.
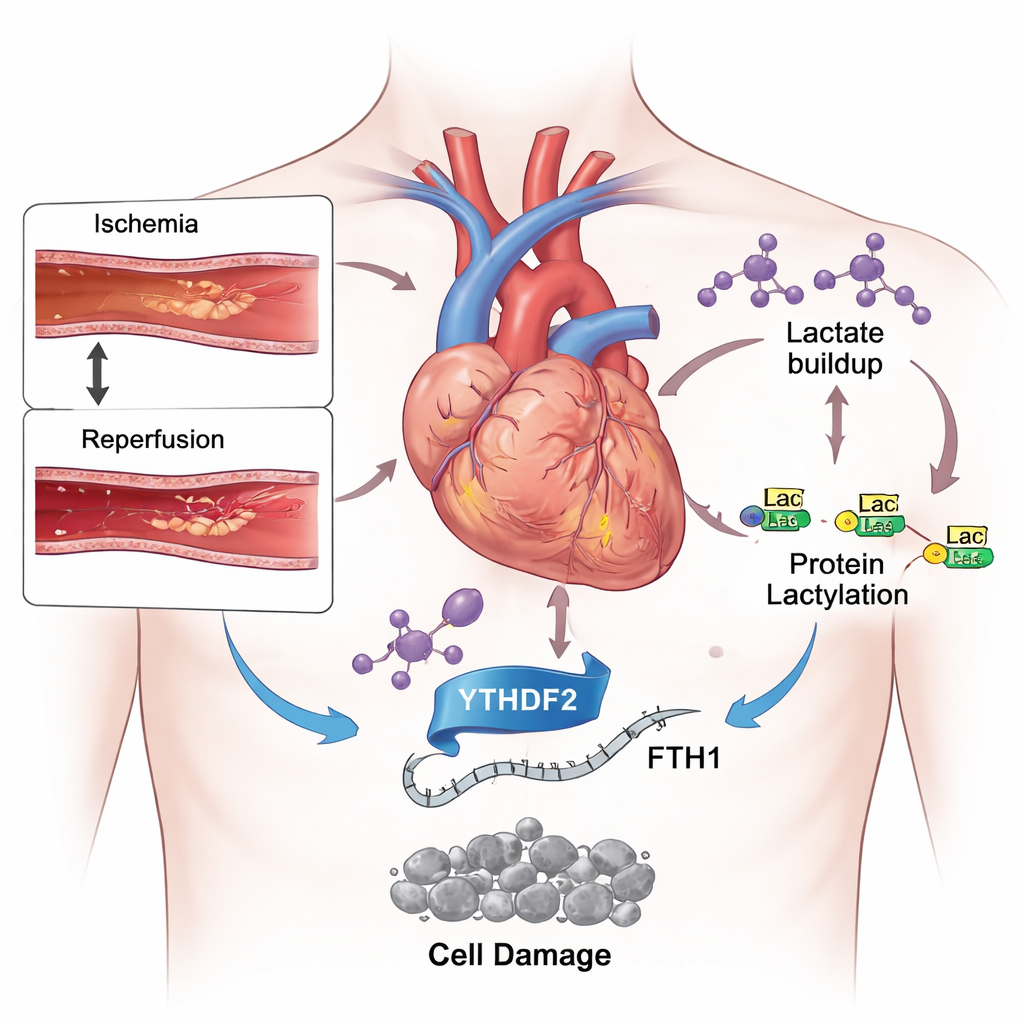
Uma espada de dois gumes no tratamento do infarto
A medicina moderna melhorou muito em reabrir rapidamente artérias coronárias obstruídas, limitando o dano inicial de um infarto. Ainda assim, pacientes podem perder grandes áreas de músculo cardíaco depois que o fluxo sanguíneo é restabelecido. Uma razão é que o retorno súbito de oxigênio e nutrientes gera uma tempestade de estresse químico dentro dos cardiomiócitos. Entre vários tipos de morte celular desencadeados nesse contexto, um mais recente chamado ferroptose atraiu atenção. Ao contrário de formas mais familiares, como apoptosis, a ferroptose depende do ferro e da oxidação desenfreada de lipídios nas membranas celulares, o que pode enfraquecer o coração de forma persistente.
Como o lactato deixa de ser apenas a “queimação muscular”
Durante um infarto, o músculo cardíaco privado de combustível desloca seu uso energético para a glicólise, um sistema de reserva que quebra o açúcar rapidamente, mas produz grandes quantidades de lactato. Usando camundongos submetidos a breve obstrução e reabertura de uma artéria coronária, e células cardíacas cultivadas expostas a baixa oxigenação e depois reoxigenação, os pesquisadores encontraram níveis de lactato fortemente aumentados. Ao mesmo tempo, detectaram mais de uma marca química chamada lactilação em muitas proteínas e em histonas, as estruturas que organizam o DNA. Quando administraram aos animais um fármaco que desacelera a glicólise e reduz a produção de lactato, o dano cardíaco diminuiu, marcadores sanguíneos de lesão caíram, e o equilíbrio entre ferro prejudicial e antioxidantes protetores melhorou. Esses resultados sugerem que o excesso de lactato não é apenas um subproduto do estresse, mas um motor ativo do dano.
Um interruptor molecular que afrouxa o controle do ferro
Indo mais a fundo, a equipe concentrou-se em YTHDF2, uma proteína que lê marcas químicas no RNA e decide com que rapidez certas mensagens são destruídas. Eles descobriram que a isquemia–reperfusão e a adição de lactato aumentaram tanto os níveis de YTHDF2 quanto a lactilação em torno do gene que o codifica, amplificando sua produção. Um dos alvos-chave de YTHDF2 revelou-se ser o RNA da cadeia pesada da ferritina 1 (FTH1), uma parte central da gaiola de armazenamento de ferro da célula. Normalmente, a FTH1 aprisiona o ferro em forma segura, impedindo que ele alimente reações danosas. Em cardiomiócitos estressados, YTHDF2 se ligou mais fortemente ao RNA de FTH1 e acelerou sua degradação, deixando as células com menos “gaiolas” de ferritina, mais ferro livre, estresse oxidativo aumentado e sinais clássicos de ferroptose.
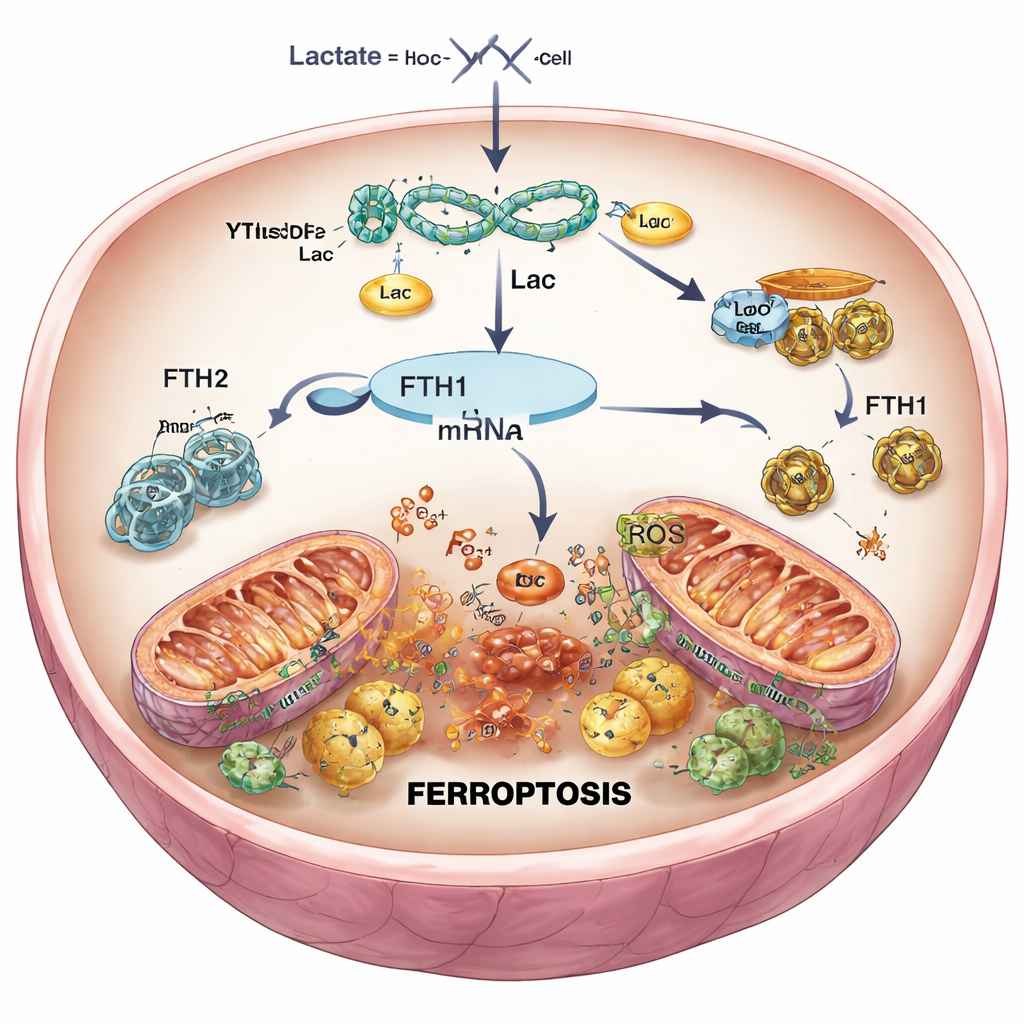
Diminuindo o sinal de morte nas células cardíacas
Para testar causa e efeito, os pesquisadores usaram ferramentas genéticas para reduzir seletivamente YTHDF2 em cardiomiócitos e em camundongos. Quando YTHDF2 foi silenciado, os níveis de FTH1 se recuperaram, ferro e espécies reativas de oxigênio diminuíram, as mitocôndrias mantiveram uma forma mais normal, e a sobrevivência celular melhorou após a reperfusão simulada. Nos camundongos, menos YTHDF2 significou cicatrizes de infarto menores e tecido com aparência mais saudável. Contudo, quando FTH1 foi simultaneamente reduzida, esses benefícios desapareceram em grande parte: o ferro aumentou novamente, o dano oxidativo retornou e o tamanho do infarto cresceu. Isso confirmou que YTHDF2 promove a ferroptose principalmente ao suprimir FTH1, afrouxando o controle do ferro dentro dos cardiomiócitos.
O que isso significa para terapias cardíacas futuras
Juntando as peças, o estudo delineia uma nova cadeia de eventos: uma artéria bloqueada e depois reaberta provoca acúmulo de lactato; o lactato eleva YTHDF2 via lactilação; YTHDF2 então destrói as instruções em RNA para a proteína guardiã do ferro FTH1; e a sobrecarga de ferro resultante desencadeia ferroptose, aprofundando o dano cardíaco. Para os pacientes, a mensagem é esperançosa: essa via oferece vários novos pontos de intervenção. Fármacos que limitem a sinalização nociva do lactato, bloqueiem a modificação específica de YTHDF2 ou preservem a função de FTH1 poderiam tornar a reperfusão de emergência mais segura e proteger mais músculo cardíaco. Embora essas descobertas ainda precisem de confirmação em tecidos humanos, elas abrem uma via promissora rumo a tratamentos mais suaves e eficazes para sobreviventes de infarto.
Citação: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Palavras-chave: infarto, lactato, morte celular dirigida por ferro, lesão por isquemia-reperfusão, proteção de cardiomiócitos