Clear Sky Science · pt
Anotação de 200 Genomas de Insetos com BRAKER para Comparações Consistentes entre Espécies
Por que os Genomas de Insetos Importam
Os insetos moldam nosso mundo: eles polinizam culturas, transmitem doenças, reciclam nutrientes e inspiram novos materiais e tecnologias. Hoje podemos ler o DNA de milhares de espécies de insetos, mas ter apenas seus genomas não é suficiente. Também precisamos de um mapa claro de onde cada gene está e qual é sua função provável. Este artigo descreve um grande esforço padronizado para anotar os genes de 200 espécies de insetos usando um fluxo de trabalho automatizado chamado VARUS-BRAKER, tornando muito mais fácil para os cientistas comparar espécies e descobrir como os insetos evoluíram sua notável diversidade.
O Problema dos Mapas Genéticos Incompletos
Nas últimas duas décadas, o sequenciamento de genomas de insetos explodiu, passando de cerca de vinte espécies para mais de quatro mil. No entanto, apenas cerca de uma em cada dez desses genomas possui uma anotação gênica adequada em bases de dados públicas. Mesmo quando há anotações, muitas foram criadas anos atrás com métodos e dados limitados. Grupos de pesquisa diferentes frequentemente usaram softwares e evidências distintas, o que pode criar diferenças artificiais: um gene pode parecer ausente ou com formato estranho em uma espécie simplesmente porque foi anotado com outra ferramenta. Esse mosaico de métodos torna arriscado tirar conclusões sobre como os genes de insetos realmente diferem entre espécies.
Um Fluxo de Trabalho "Um Clique" para Muitas Espécies
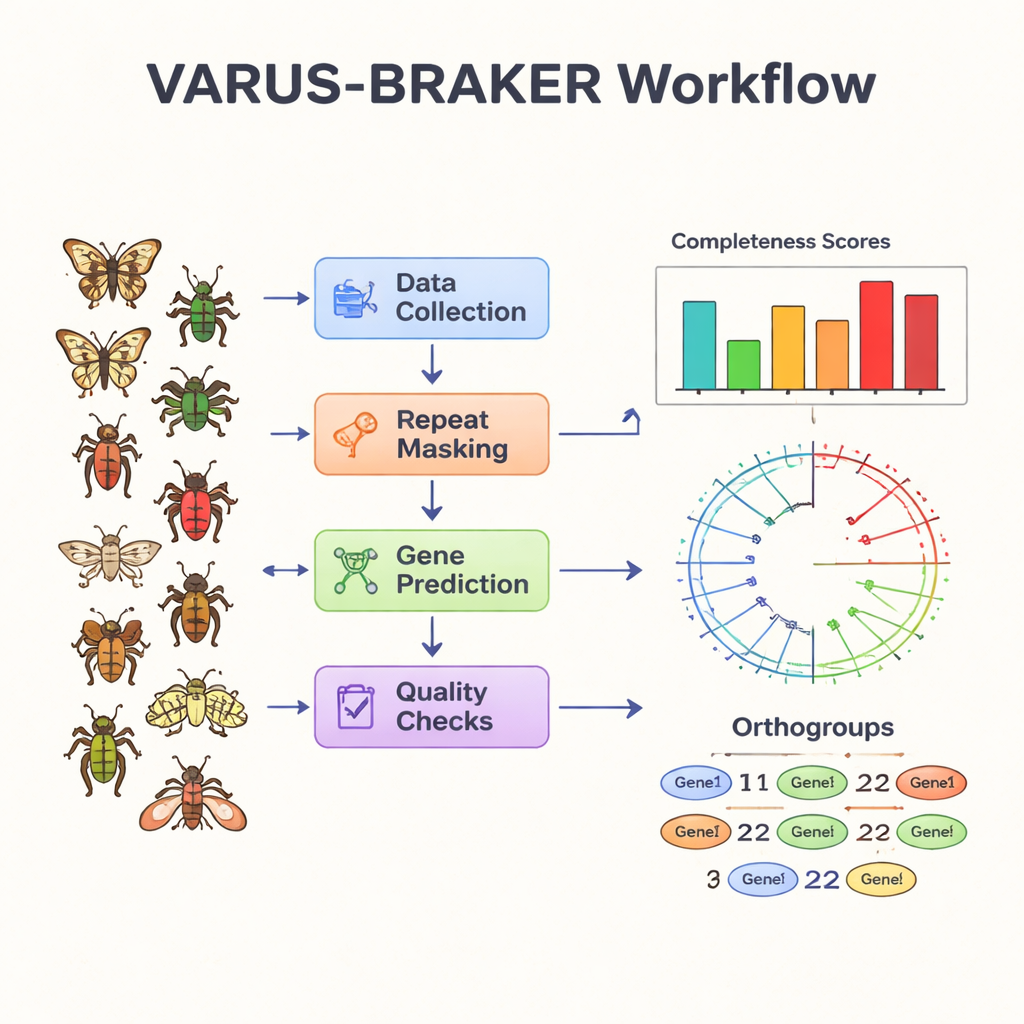
Os autores enfrentam esse gargalo construindo um fluxo de trabalho automatizado centrado no pipeline de predição gênica BRAKER3. O sistema VARUS-BRAKER foi projetado de modo que, no modo mais simples, o usuário precise fornecer apenas o nome científico de uma espécie. O fluxo de trabalho então baixa automaticamente o melhor genoma disponível em repositórios públicos, coleta dados de sequenciamento de RNA correspondentes que mostram quais genes estão ativos e recupera informações de proteínas de espécies relacionadas. Ele mascara DNAs repetitivos, alinha leituras de RNA ao genoma e combina as "pistas" vindas de RNA e proteínas para ensinar aos modelos onde os genes provavelmente começam, terminam e se emendam. Verificações de qualidade como BUSCO e OMArk avaliam quão completo e limpo é o conjunto gênico resultante.
Um Amplo Percurso pela Árvore dos Insetos
Usando esse sistema, a equipe anotou 200 genomas de insetos escolhidos para cobrir os principais ramos da árvore filogenética dos insetos, com foco em insetos holometábolos — aqueles com metamorfose completa de larva para pupa e adulto — além de um conjunto diverso de parentes. A amostra abrange 77 famílias e 14 ordens, incluindo moscas, borboletas, besouros, abelhas, formigas, pulgões, baratas e outros. Oitenta e cinco dessas espécies não tinham anotação prévia no GenBank. Para cada espécie, o fluxo de trabalho previu genes codificadores de proteínas, resultando em mais de 4,2 milhões de sequências proteicas. A maioria dos genomas e seus proteomas previstos passou por testes rigorosos de completude, tipicamente alcançando entre 85% e 95% de cobertura dos genes essenciais esperados, indicando que a abordagem automatizada produz resultados de alta qualidade.
De Listas de Genes ao Significado Biológico
Listar genes é apenas parte da história; os pesquisadores também precisam de pistas sobre o que esses genes fazem. Com esse objetivo, os autores aplicaram um pipeline de anotação funcional chamado FANTASIA, que usa modelos atuais de linguagem para proteínas para atribuir termos da Gene Ontology (GO) — rótulos padrão para funções biológicas — a cada proteína. Em comparação com a ferramenta amplamente usada InterProScan, o FANTASIA anotou cerca de 1,6 vez mais proteínas, mantendo concordância próxima quando ambos os métodos faziam predições. A equipe também agrupou genes relacionados em "ortogrupos", conjuntos de genes que compartilham um ancestral comum, e usou esses grupos para construir uma árvore evolutiva das 200 espécies. Essa estrutura permite investigar quais genes são compartilhados, perdidos ou expandidos em diferentes linhagens de insetos, e conectar repertórios gênicos a características como metamorfose ou comportamento larval.
Um Recurso Reutilizável para Descobertas Futuras
Todos os dados deste projeto — incluindo estruturas gênicas, sequências de proteínas, rótulos funcionais, ortogrupos, árvores de espécies e predições de tRNA — estão disponíveis gratuitamente em repositórios públicos. Os autores também publicam o fluxo completo VARUS-BRAKER como código open-source para que outros cientistas possam anotar novos genomas de insetos, ou mesmo de outros animais e plantas, de maneira consistente. Para não especialistas, a principal conclusão é que este trabalho transforma uma coleção dispersa de sequências de DNA em um atlas coerente e comparável de genes de insetos. Com esses mapas padronizados, estudos futuros podem descobrir com mais segurança como os insetos evoluíram o voo, a metamorfose e o sucesso ecológico, e podem identificar com maior precisão genes relevantes para agricultura, conservação e controle de doenças.
Citação: Saenko, S., Hoff, K.J. & Stanke, M. Annotation of 200 Insect Genomes with BRAKER for Consistent Comparisons across Species. Sci Data 13, 288 (2026). https://doi.org/10.1038/s41597-026-06840-0
Palavras-chave: genômica de insetos, anotação de genoma, genômica comparativa, biologia evolutiva, bioinformática