Clear Sky Science · pt
Conjunto de dados virtual mínimo para montagem de genoma de novo triploide reprodutível
Por que genomas com três cópias importam
Muitas culturas e outros organismos não carregam apenas duas cópias de cada cromossomo, como os humanos—eles podem ter três ou mais. Montar essas cópias extras a partir de dados de sequenciamento de DNA é surpreendentemente difícil, porque as cópias são muito parecidas, mas não idênticas. Este artigo apresenta um conjunto de dados “virtual” pequeno, porém cuidadosamente projetado, que permite aos pesquisadores testar e comparar softwares de montagem de genoma em um problema realista de três cópias (triploide), sob condições completamente conhecidas e reprodutíveis.
Construindo um genoma substituto simples
Em vez de começar a partir de uma planta ou animal real, o autor cria primeiro um trecho aleatório de DNA de um milhão de letras para servir como um molde limpo. Esse molde é então duplicado em três versões separadas, representando os três conjuntos de cromossomos em um organismo triploide. Para imitar como genomas reais mudam lentamente ao longo do tempo, o estudo introduz um número fixo de pequenas alterações—substituições de uma única letra—passo a passo em cada cópia. Repetir esse processo por 100 passos produz tríades de genomas que vão de quase idênticos a claramente, mas moderadamente, diferentes. Esse “gradiente de divergência” controlado é a espinha dorsal do benchmark.
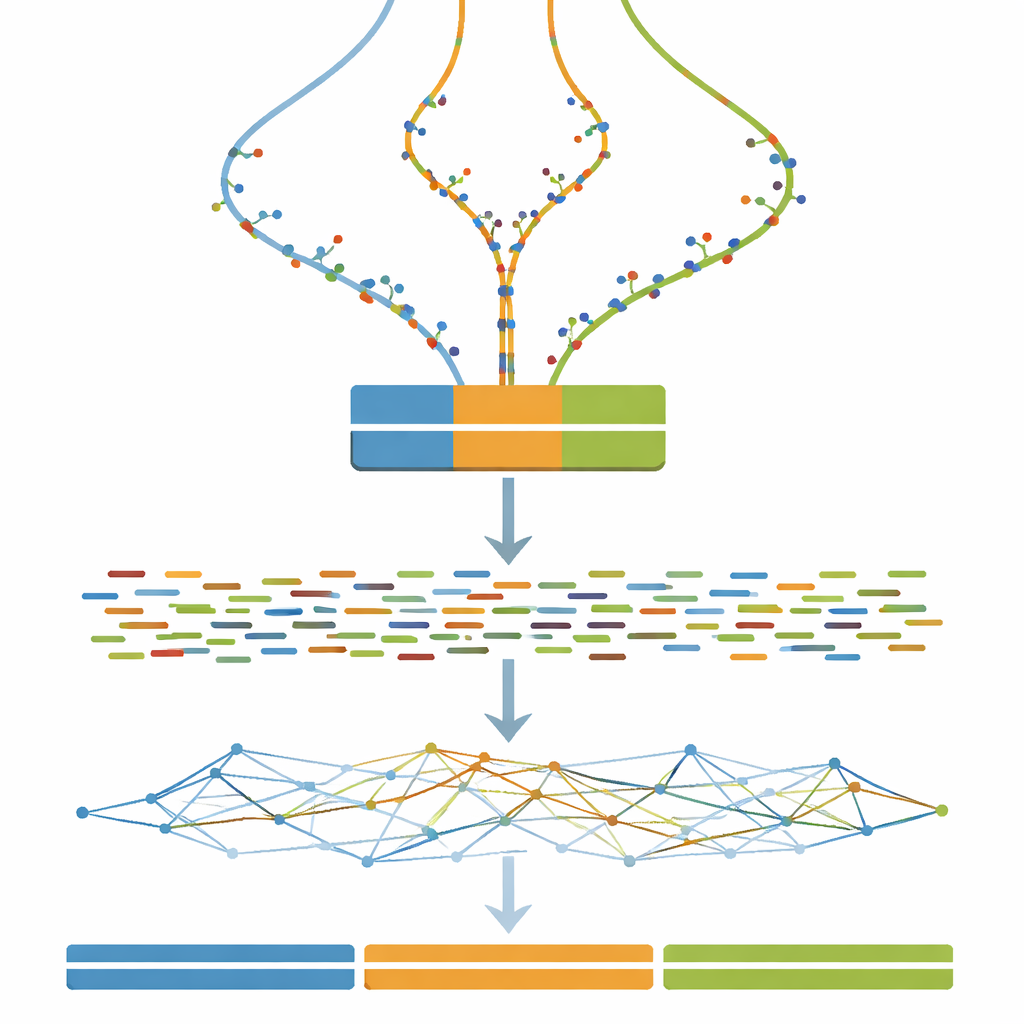
Transformando genomas virtuais em experimentos virtuais
Uma vez definido cada genoma de três cópias, o próximo passo é imitar o que uma máquina de sequenciamento de DNA veria. O estudo usa software amplamente adotado para simular fragmentos curtos de DNA pareados, semelhantes aos produzidos por um sequenciador Illumina, em uma profundidade de cobertura constante e relativamente alta. Passos opcionais de limpeza imitam práticas comuns do mundo real, como corrigir erros randômicos de sequenciamento e mesclar pares de reads que se sobrepõem. Como resultado, qualquer pessoa que use o conjunto de dados pode testar não apenas seus algoritmos de montagem, mas também como escolhas típicas de pré-processamento influenciam os genomas montados finais.
Testando sob estresse estratégias de montagem
O cerne do trabalho é um grande experimento no qual todos os reads simulados são alimentados em um único programa de montagem de genoma enquanto se altera apenas uma configuração chave: o tamanho do k-mer, um parâmetro que controla quão finamente o software “fatia” os reads ao reconstruir o genoma. Para cada combinação de nível de divergência (de 0 a 100 passos) e tamanho de k-mer (uma ampla faixa de valores ímpares), uma nova montagem é construída. Uma ferramenta de avaliação acompanhante mede então quão contínuas são as peças montadas, quantas peças existem e quão próxima sua soma de comprimentos está da verdade conhecida de três milhões de letras. Essas medidas são resumidas em mapas de calor, revelando amplas zonas onde as montagens colapsam diferentes cópias em uma só, fragmentam em muitas peças pequenas ou se aproximam do ideal de três contigs longos e precisos.
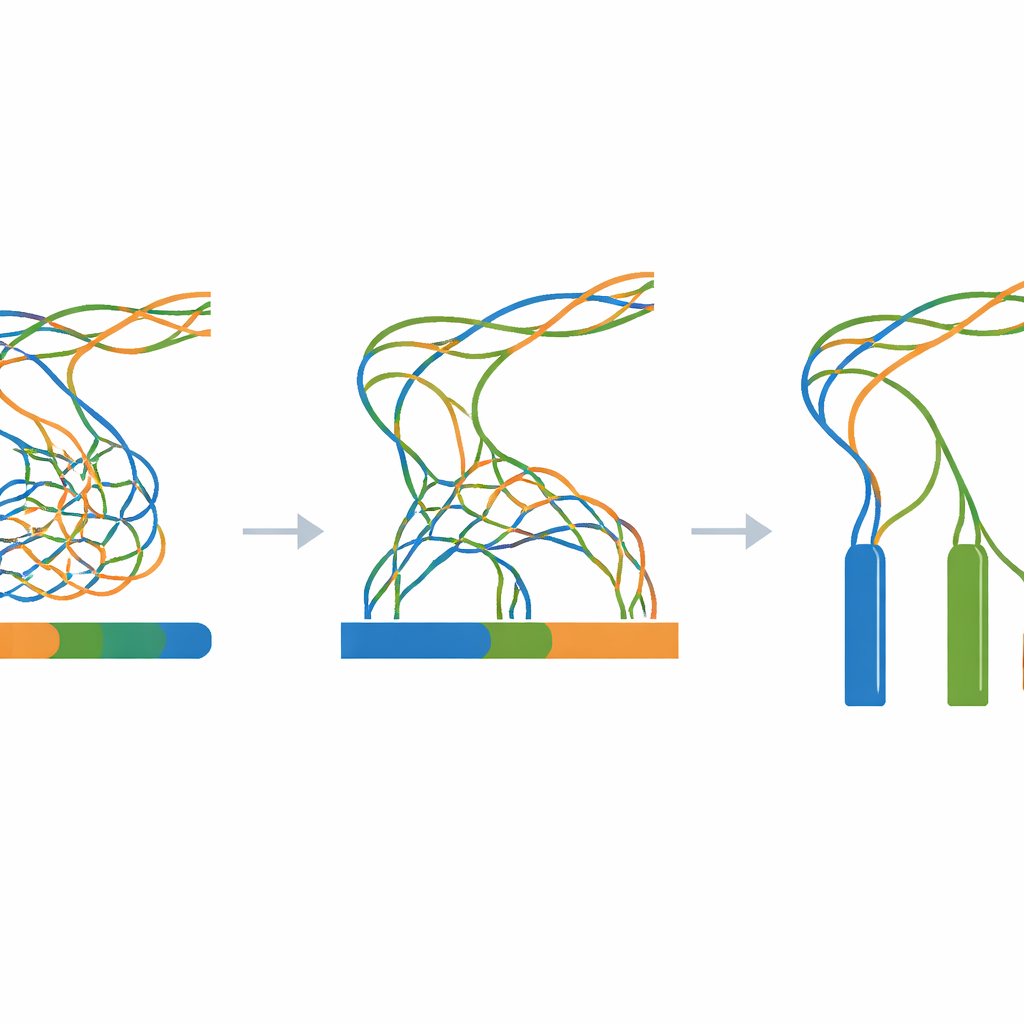
Uma referência transparente para genomas complicados
Como cada etapa é sintética e roteirizada—desde o molde aleatório inicial até as montagens finais—os pesquisadores podem reproduzir todo o fluxo de trabalho em qualquer computador Linux padrão usando apenas ferramentas de código aberto. O arquivo no Zenodo vinculado ao artigo contém o genoma molde, todas as sequências intermediárias mutadas, todos os reads simulados e cada resultado de montagem, junto com logs e scripts auxiliares simples. Verificações técnicas confirmam que o processo de mutação se comporta conforme esperado, que os reads simulados correspondem aos comprimentos e cobertura solicitados e que as montagens exibem o padrão antecipado: forte colapso excessivo quando as três cópias são quase idênticas e separação mais clara à medida que elas se distanciam.
O que isso significa em termos simples
Em linguagem cotidiana, este artigo oferece uma pista de testes controlada para software que tenta reconstruir três livros de instruções semelhantes a partir de pilhas de fragmentos embaralhados. Ao aumentar gradualmente o quanto os três livros diferem e ao alterar sistematicamente uma configuração chave no processo de reconstrução, o conjunto de dados facilita ver quando e como os métodos atuais falham ou têm sucesso. Desenvolvedores podem usá‑lo para ajustar novos algoritmos, enquanto usuários podem entender melhor quais configurações funcionam melhor para genomas triploides. Embora o DNA em si seja artificial, as lições que permite—sobre colapso, separação e o impacto das escolhas de parâmetros—são diretamente relevantes para esforços do mundo real de decodificar os genomas complexos de muitas espécies importantes.
Citação: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Palavras-chave: montagem de genoma triploide, avaliação de poliploidia, conjunto de dados de DNA sintético, montagem de novo, otimização de k-mer