Clear Sky Science · pt
AF2BIND: previsão de sítios de ligação a pequenas moléculas usando a representação de pares do AlphaFold2
Encontrando alvos farmacológicos em um mar de proteínas
Medicamentos modernos frequentemente atuam se fixando em pequenas reentrâncias nas superfícies das proteínas dentro das nossas células. Mesmo com os enormes catálogos atuais de estruturas proteicas, continua surpreendentemente difícil prever de antemão onde uma pequena molécula — um potencial fármaco — poderá realmente aderir. Este estudo apresenta o AF2BIND, uma ferramenta computacional simples, porém poderosa, que explora os mecanismos internos do AlphaFold2, o marco na predição de estruturas de proteínas, para destacar prováveis sítios de ligação a fármacos em milhares de proteínas humanas. Seu objetivo é restringir a busca por novos medicamentos e revelar pontos funcionais ocultos que métodos tradicionais deixam passar.
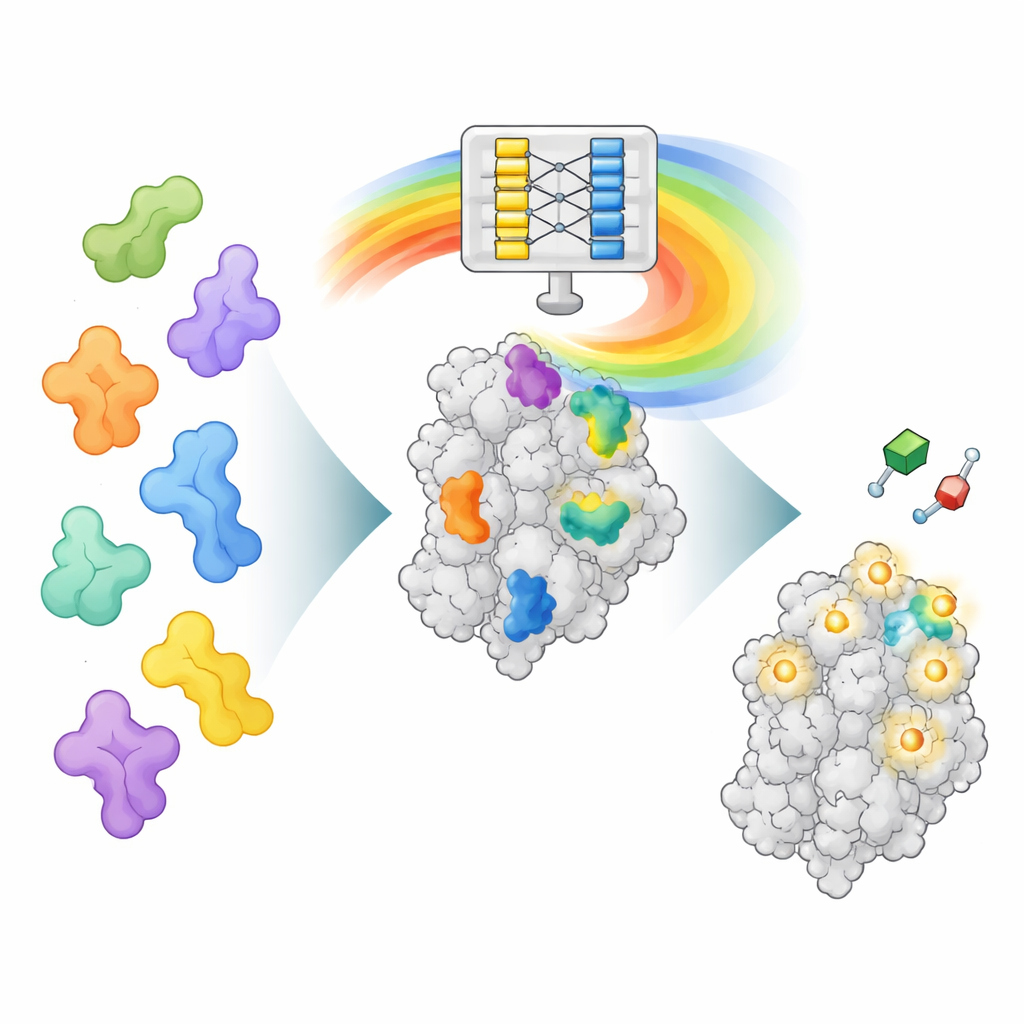
Uma nova maneira de ler a “mente” do AlphaFold
O AlphaFold2 foi treinado para prever como uma cadeia de aminoácidos se dobra em uma proteína tridimensional, não para localizar onde fármacos se ligam. Contudo, ao aprender o dobramento, ele também assimilou padrões ricos sobre como diferentes partes da proteína interagem. O AF2BIND aproveita uma dessas camadas de dados internas, chamada representação de pares, que codifica como cada par de posições de aminoácidos se relaciona no espaço. Os autores fornecem ao AlphaFold2 uma sequência proteica junto com sua estrutura da espinha dorsal e ainda acrescentam 20 aminoácidos extras, um de cada tipo, como cadeias “iscas” separadas. O AlphaFold2 então calcula como a proteína interage com cada resíduo isca. Esses padrões de interação viram a entrada para um modelo de regressão logística muito direto que estima, para cada posição na proteína, a probabilidade de pertencer a um sítio de ligação a pequenas moléculas.
Transformando sinais ocultos em previsões práticas
O treinamento do AF2BIND exigiu um conjunto cuidadosamente curado de cerca de 1.900 estruturas proteína–ligante nas quais pequenas moléculas estavam ligadas com evidência experimental de alta qualidade. Os pesquisadores tomaram grandes cuidados para evitar “fraudes” por similaridade: dividiram os dados de modo que proteínas de teste não compartilhassem o dobramento geral, a sequência ou mesmo a forma do bolso de ligação com aquelas usadas no treino. Nesse benchmark rigoroso, a representação de pares do AF2 superou várias incorporações alternativas de redes neurais, incluindo as baseadas apenas em sequência ou em desenho de sequência condicionado à estrutura. Usando apenas as características de pares, o AF2BIND recuperou aproximadamente dois terços dos resíduos de ligação conhecidos entre as predições mais bem classificadas e apresentou desempenho forte em métricas padrão de classificação, mantendo robustez diante de mudanças modestas na forma da proteína e na orientação de cadeias laterais.
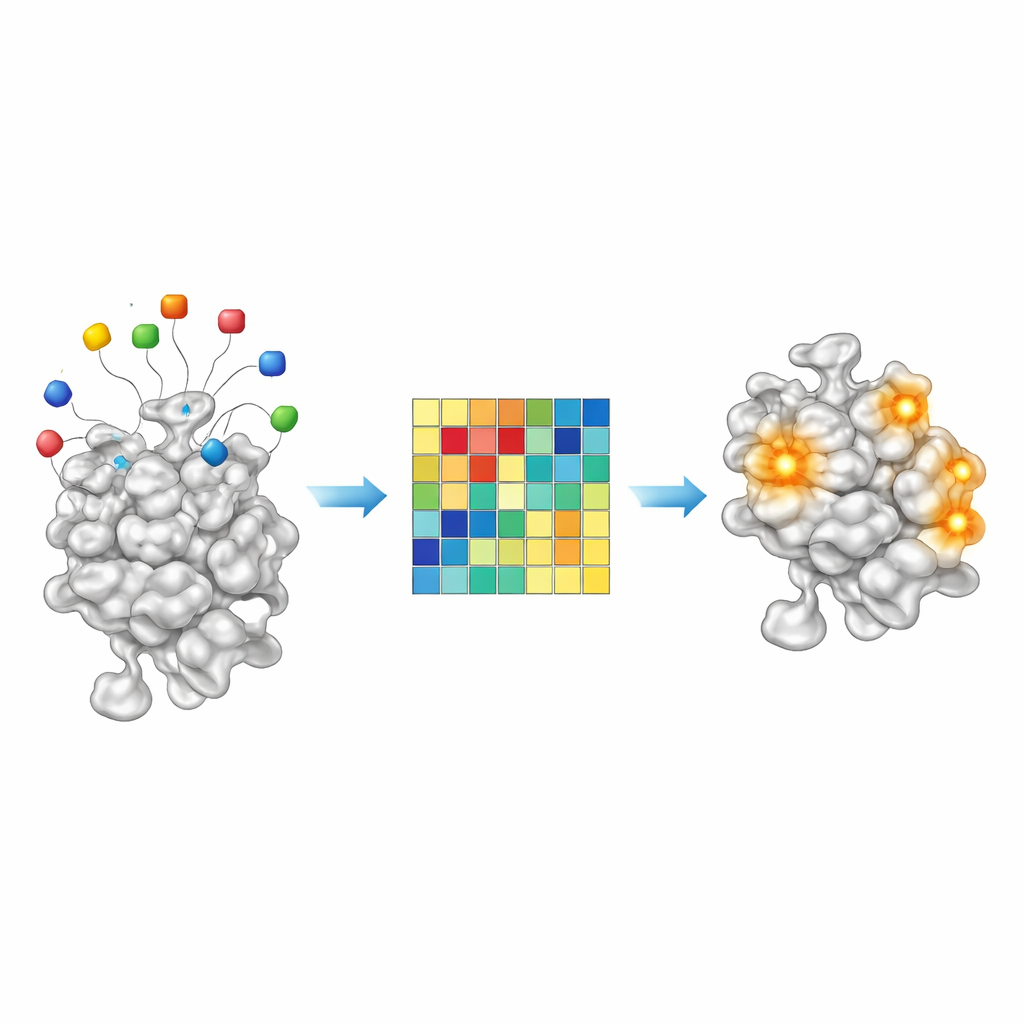
Lendo pistas químicas das residências isca
Como o AF2BIND é um modelo linear simples, suas decisões são incomumente transparentes para um sistema moderno de IA. Cada um dos 20 aminoácidos isca contribui com um valor mensurável para a pontuação final de ligação em uma dada posição da proteína. Ao examinar essas contribuições em cerca de 2.000 complexos proteína–ligante, os autores descobriram que certas combinações de iscas ativam mais fortemente para ligantes oleosos, ricos em carbono, enquanto outras se acendem para moléculas mais polares e hidrofílicas. Em outras palavras, o padrão de ativação das iscas age como uma impressão digital química rudimentar dos tipos de pequenas moléculas que um determinado bolso prefere. Isso sugere que, no futuro, abordagens do tipo AF2BIND podem não apenas indicar onde um fármaco poderia se ligar, mas também sugerir o tipo de química que se ajustaria melhor.
Escaneando o proteoma humano em busca de novos bolsos
Munidos de seu modelo treinado, a equipe então aplicou o AF2BIND às estruturas previstas pelo AlphaFold para todo o proteoma humano. Após remover regiões de baixa confiança e dividir proteínas muito grandes em pedaços estruturais manejáveis, eles agruparam resíduos de alta pontuação próximos em sítios candidatos de ligação. O AF2BIND previu mais de 20.000 desses sítios em mais de 13.000 proteínas. De forma marcante, a maioria não coincidiu com bolsos inferidos por métodos baseados em homologia, como o AlphaFill, que copia ligantes de estruturas cristalinas relacionadas, nem com um reconhecido localizador de bolsos chamado P2Rank. Muitos sítios detectados apenas pelo AF2BIND são mais rasos ou mais difusos do que os bolsos clássicos enterrados e frequentemente coincidem com regiões que ligam peptídeos, RNA, DNA ou outras proteínas — interfaces que, ainda assim, podem ser atacáveis por pequenas moléculas.
Implicações para descoberta de fármacos e doenças
Para avaliar quão promissores esses sítios recém-sugeridos poderiam ser para o desenho de fármacos, os autores usaram uma ferramenta independente que pontua a “druggabilidade” com base no tamanho do bolso, no grau de fechamento e no ambiente químico. Em média, os sítios do AF2BIND pontuaram acima de um limiar comum para alvos farmacológicos atraentes, incluindo aqueles encontrados em proteínas ligadas a doenças hereditárias. Quando cruzados com experimentos quimioproteômicos que marcam cisteínas reativas em células, o AF2BIND e o P2Rank juntos explicaram quase metade das regiões observadas como ligáveis, cada método capturando casos que o outro não detectou. O trabalho demonstra que as representações internas aprendidas por redes de predição estrutural podem ser reaproveitadas para mapear em larga escala sítios prováveis de ligação a fármacos, sem conhecimento prévio de qualquer ligante específico. Para não especialistas, a mensagem principal é que os mesmos avanços em IA que prevêem formas de proteínas começam a revelar onde e como medicamentos podem melhor “agarrar” essas formas, potencialmente acelerando a busca por novos tratamentos e iluminando pontos de controle antes ocultos em nossas proteínas.
Citação: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Palavras-chave: sítios de ligação de proteínas, descoberta de fármacos, AlphaFold2, biologia computacional, bioinformática estrutural