Clear Sky Science · pt
DECODE: estrutura comum de desconvolução baseada em deep learning para vários dados ômicos
Por que esta pesquisa importa
A biomedicina moderna está repleta de medições dos nossos tecidos: quais genes estão ativos, quais proteínas estão presentes e quais pequenas moléculas alimentam nossas células. Ainda assim, a maioria dessas medições é feita em amostras misturadas, onde muitos tipos celulares estão combinados. O estudo por trás do DECODE apresenta uma poderosa estrutura de inteligência artificial que pode separar esses sinais, indicando quais células e estados celulares estão presentes, mesmo em tipos de dados muito diferentes. Essa capacidade pode acelerar pesquisas sobre câncer, imunidade e doenças metabólicas, além de aproveitar melhor amostras já armazenadas em biobancos.
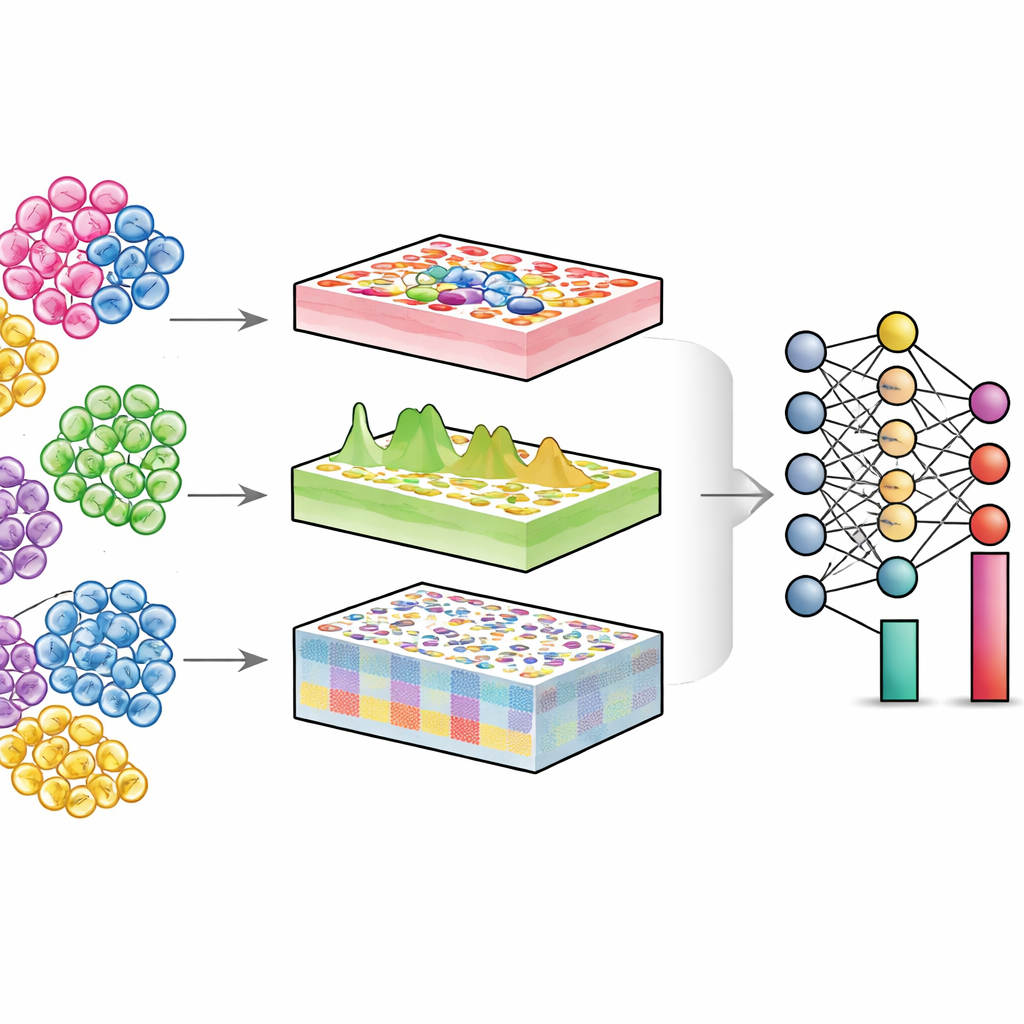
Investigando tecidos mistos
Cada órgão é uma comunidade de diferentes tipos celulares—células do sistema imune, células estruturais, células‑tronco e outras. Em saúde e doença, o que frequentemente muda não é apenas o que cada célula faz, mas quantas de cada tipo estão presentes e em que estado se encontram. Tecnologias de célula única podem medir células individuais diretamente, mas são caras e tecnicamente desafiadoras, especialmente para grandes coortes de pacientes ou amostras antigas armazenadas. Em contraste, experimentos convencionais em “bulk” misturam milhares ou milhões de células e fornecem um sinal médio. Algoritmos de desconvolução tentam reverter essa mistura: dados os dados bulk e um mapa de referência de células únicas, eles estimam a proporção de cada tipo celular no tecido.
Os limites de ferramentas especializadas
Ferramentas de desconvolução existentes costumam ser adaptadas a um único tipo de medição, como atividade gênica (transcriptômica) ou proteínas (proteômica). Frequentemente assumem comportamentos estatísticos específicos que não se aplicam a outros tipos de dados e têm dificuldades quando o tecido bulk contém tipos celulares ausentes na referência. Fortes efeitos de batch—diferenças entre doadores, instrumentos ou estados de saúde—podem ainda mais borrar os sinais biológicos. Notavelmente, não havia um método prático para metabolômica, o estudo de pequenas moléculas que muitas vezes estão mais próximas dos sintomas clínicos. Como resultado, cientistas que analisavam coortes multiómicas precisavam conciliar várias ferramentas especializadas, cada uma com suas peculiaridades, tornando difícil comparar resultados entre estudos e tipos de dados.
Um motor universal de separação
O DECODE enfrenta esses desafios tratando a desconvolução como um problema flexível de deep learning que pode lidar com genes, proteínas e metabólitos de forma unificada. Primeiro, ele sintetiza “pseudotecidos” misturando digitalmente perfis de célula única em proporções aleatórias, criando um conjunto de treinamento rico onde a composição celular verdadeira é conhecida. Uma etapa de aprendizado adversarial então treina um codificador para mapear tecidos reais e pseudotecidos em uma representação compartilhada, na qual diferenças técnicas são minimizadas, mas padrões biologicamente significativos são preservados. Em seguida, um módulo especial de denoising, guiado por aprendizado contrastivo, aprende a separar sinais verdadeiros do tecido de ruídos artificiais. Essa etapa torna o DECODE robusto a tipos celulares em falta na referência e a erros de medição. Finalmente, as características limpas são encaminhadas a um módulo de desconvolução que estima abundâncias absolutas ou relativas de tipos e estados celulares, dependendo de quão completa é a referência.
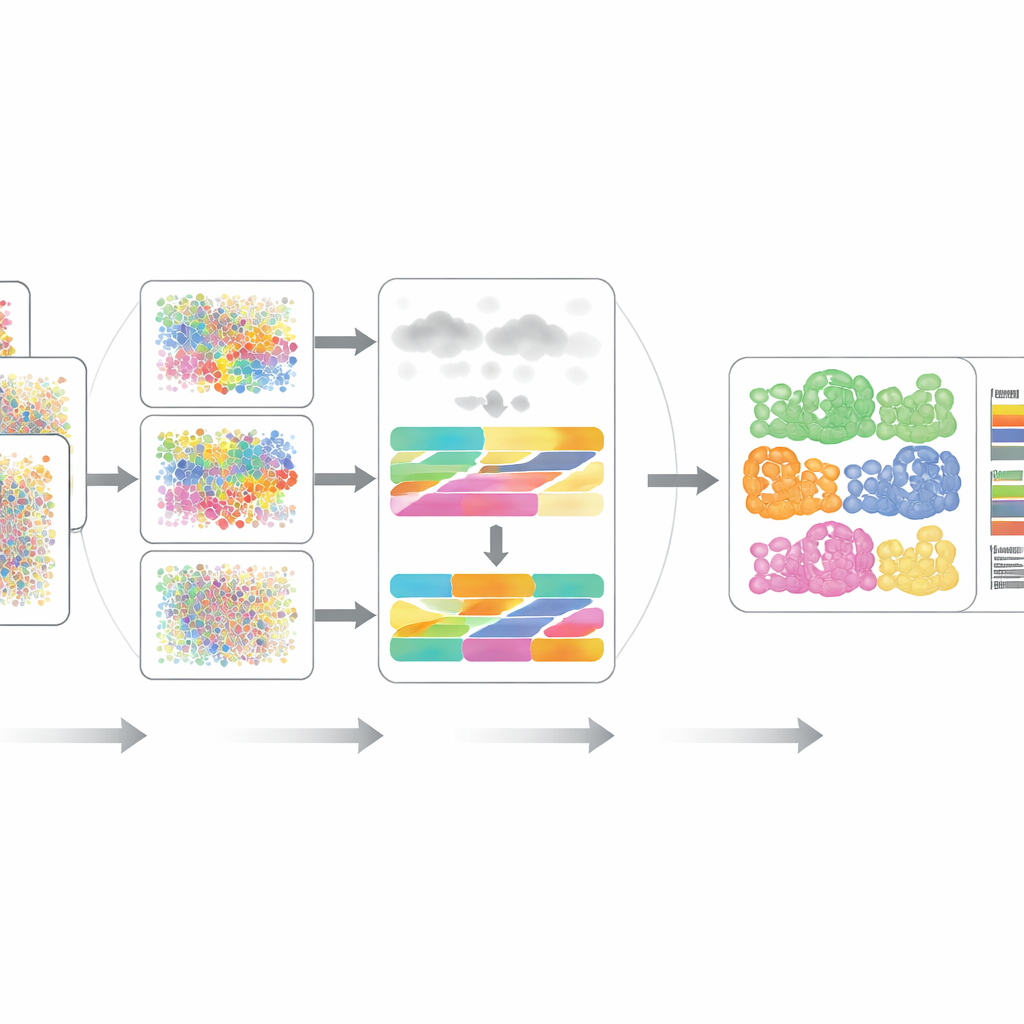
Colocando o DECODE à prova
Os autores avaliaram rigorosamente o DECODE em 15 conjuntos de dados que cobrem sete cenários realistas, incluindo diferentes doadores, estados de doença, condições de saúde, plataformas experimentais e até medições com resolução espacial. Em transcriptômica e proteômica, o DECODE geralmente igualou ou superou ferramentas de ponta em precisão, mantendo tempo de computação e uso de memória razoáveis. Crucialmente, o DECODE foi o único método a entregar resultados confiáveis em dados de metabolômica, onde há menos features e tipos celulares diferentes podem parecer enganadoramente semelhantes. A estrutura também se mostrou competente em rastrear estados celulares—como progressão ao longo de uma trajetória de desenvolvimento, fases do ciclo celular ou respostas a tratamentos—e não apenas tipos celulares estáticos.
Robusto em dados do mundo real, ruidosos e incompletos
Tecidos reais frequentemente contêm tipos celulares não capturados em referências experimentais de célula única, e o ruído experimental pode distorcer muitas características ao mesmo tempo. Os pesquisadores simularam esses problemas adicionando tipos celulares desconhecidos e introduzindo várias formas de ruído e dados faltantes em transcriptômica, proteômica e metabolômica. Na maioria dos cenários, o DECODE permaneceu o método mais preciso e, em metabolômica, o único que não entrou em colapso. Eles também demonstraram que o DECODE fornece respostas altamente consistentes quando aplicado a medições emparelhadas de genes e proteínas das mesmas amostras de células sanguíneas, um requisito chave para comparar mudanças de tipos celulares entre camadas ômicas em grandes coortes.
Novos insights biológicos a partir de coortes multiómicas
Munidos dessa ferramenta unificada, a equipe revisitou conjuntos de dados de doenças complexas. No câncer de mama, eles combinaram coortes transcriptômicas e proteômicas para mostrar como células imunológicas e células estromais de suporte mudam entre tumores não metastáticos, tumores primários metastizantes e metástases cerebrais. Padrões como maior abundância de células T e células semelhantes a perivasculares em lesões não metastáticas, e aumento de células B em doença avançada, corroboram e ampliam estudos biológicos prévios. No fígado de camundongo, o DECODE integrou coortes transcriptômicas, proteômicas e metabolômicas para acompanhar como hepatócitos, células endoteliais e células imunológicas residentes mudam sob diferentes dietas e modelos de doença hepática, reproduzindo tendências conhecidas, como aumento da fração de células de Kupffer em condições inflamatórias.
O que isso significa daqui para frente
Para um leitor leigo, a mensagem principal é que o DECODE age como um prisma inteligente para dados biomédicos: dadas medições misturadas de tecidos, ele consegue separar as contribuições de muitos tipos e estados celulares, e o faz de forma confiável em vários tipos de leitura molecular. Isso permite que cientistas extraiam muito mais informação de coortes multiómicas e biobancos existentes sem coletar novos dados de célula única para cada projeto. Embora o método ainda dependa da qualidade e abrangência das referências disponíveis de célula única, e recursos de metabolômica permaneçam limitados, o DECODE marca um avanço significativo rumo à interpretação rotineira em nível celular de estudos humanos em larga escala, com benefícios potenciais para entender mecanismos de doença e orientar a medicina de precisão.
Citação: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Palavras-chave: desconvolução multiómica, referência de célula única, deep learning em biologia, análise de metabolômica, composição de tipos celulares