Clear Sky Science · pt
Variação de taxa e erros recorrentes em sequências na filogenética em escala pandêmica
Por que isso importa para surtos futuros
Quando um novo vírus se espalha pelo mundo, cientistas correm para ler seu código genético e reconstruir sua árvore genealógica. Essas árvores ajudam a rastrear como surgem variantes, com que rapidez se espalham e se as medidas de controle funcionam. Mas durante a COVID-19, laboratórios sequenciaram milhões de genomas de SARS‑CoV‑2 tão rapidamente que erros e peculiaridades ocultas nos dados começaram a distorcer a imagem. Este artigo apresenta novos métodos para limpar e interpretar conjuntos de dados genéticos tão vastos, oferecendo visões mais claras de como um vírus pandêmico realmente evolui e se movimenta entre populações.
O desafio de interpretar milhões de genomas
A epidemiologia genômica transforma genomas virais em informação prática para decisões de saúde pública. Para o SARS‑CoV‑2, mais de 20 milhões de genomas foram compartilhados mundialmente. Ferramentas evolutivas tradicionais foram desenvolvidas para problemas mais modestos, como comparar genes entre espécies, e não para lidar com milhões de sequências virais quase idênticas chegando em tempo real. Nessa escala, dois problemas tornam-se especialmente problemáticos. Primeiro, alguns sítios no genoma viral mutam com muito mais frequência que outros, o que pode fazer vírus não relacionados parecerem estranhamente semelhantes. Segundo, erros técnicos recorrentes no sequenciamento e no processamento de dados podem imitar mutações reais. Ambos os efeitos geram “ecos falsos” na árvore evolutiva, criando incerteza sobre quais ramos e agrupamentos confiar.
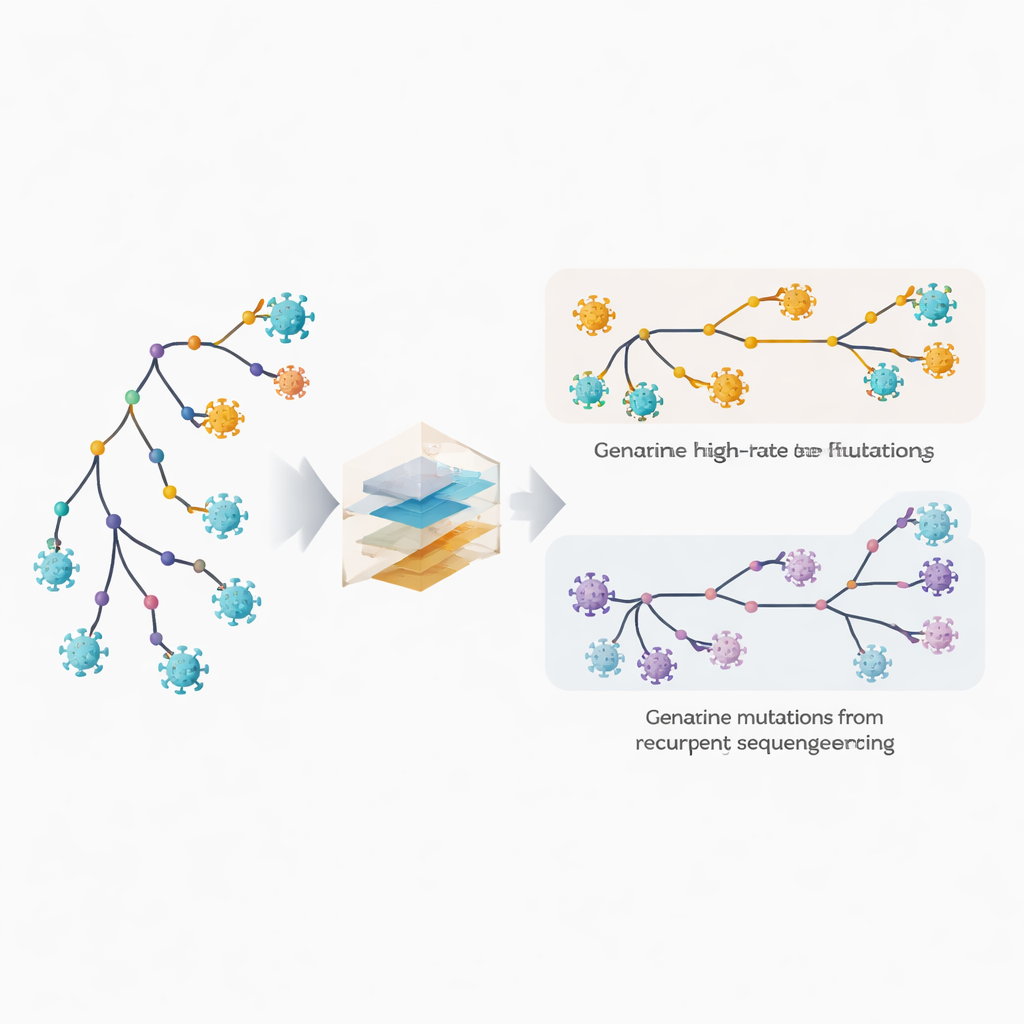
Detectando sítios de mutação rápida e erros ocultos
Os autores estendem seu software filogenético, MAPLE, com modelos que tratam cada posição no genoma viral como tendo seu próprio comportamento. Em vez de assumir algumas taxas médias de mutação, o método estima uma taxa separada para cada sítio, aproveitando o enorme número de genomas disponíveis. Ao mesmo tempo, permite que cada sítio tenha sua própria probabilidade de carregar um erro recorrente de sequenciamento ou de chamada de consenso. O truque principal é comparar com que frequência uma alteração aparece em ramos internos profundos da árvore, que refletem eventos compartilhados mais antigos, versus nas pontas externas, que correspondem a genomas individuais. Mutações biológicas verdadeiras tendem a estar distribuídas entre ramos internos e terminais, enquanto erros técnicos aparecem sobretudo nas pontas. Explorando esse padrão, o método consegue desvincular rápida evolução genuína de erros repetidos.
Algoritmos mais rápidos para uma árvore da vida lotada
Lidar com milhões de genomas normalmente exigiria poder computacional enorme. Para manter a análise prática, a equipe redesenhou como o MAPLE armazena e atualiza informações de sequência na árvore. Em vez de comparar cada genoma com uma única referência fixa, o software seleciona pontos de “referência local” dentro da árvore e registra genomas próximos como diferenças relativas a esses pontos âncora. Essa representação compacta acelera comparações entre partes distantes da árvore. Melhorias adicionais refinam como novas amostras são adicionadas a uma árvore existente, como os comprimentos de ramo são ajustados e como formas alternativas da árvore são exploradas, com opções para executar as etapas mais exigentes em paralelo em múltiplos núcleos de processamento.
Testando o método e limpando dados do mundo real
Para verificar se seus modelos funcionam, os autores primeiro criaram conjuntos de dados simulados realistas do SARS‑CoV‑2 com padrões de mutação conhecidos e erros de sequência incorporados. Nessas provas, a nova abordagem recuperou árvores evolutivas mais fiéis e localizou erros individuais com alta precisão, especialmente quando dezenas de milhares de genomas ou mais foram incluídos. Em seguida, voltaram-se para dados reais, analisando milhões de sequências de SARS‑CoV‑2 para as quais leituras brutas estavam disponíveis. Ao comparar dois diferentes pipelines de construção de consenso, identificaram posições específicas do genoma repetidamente afetadas por artefatos, como problemas de ligação de primers ou chamadas viesadas pela referência. Esses sítios suspeitos foram mascarados para análises subsequentes, e genomas que mostravam sinais de contaminação ou infecção mista foram filtrados, produzindo um alinhamento curado de mais de dois milhões de sequências de alta qualidade.
Uma imagem global mais clara da árvore familiar do vírus
Usando o conjunto de dados limpo, os autores reconstruíram uma árvore filogenética global do SARS‑CoV‑2 e mapearam como as principais variantes se relacionam entre si. Sua árvore às vezes propõe relações sutilmente diferentes das árvores públicas anteriores, frequentemente de maneiras que requerem menos eventos de mutação e se ajustam melhor ao modelo estatístico. A estrutura também destaca onde rótulos de linhagem podem ser inconsistentes com a história genética subjacente, sinalizando possíveis recombinantes ou genomas problemáticos para inspeção mais cuidadosa. Embora alguns desafios permaneçam — como overfitting quando os dados são escassos, ou a influência de amostras fortemente contaminadas — o trabalho mostra que agora é viável construir árvores evolutivas em escala pandêmica mais confiáveis. Para um leitor leigo, a conclusão é que um melhor tratamento de erros e pontos quentes de mutação leva a uma compreensão mais nítida de como patógenos se espalham e mudam, ajudando cientistas e agências de saúde a responderem mais rápida e seguramente em surtos futuros.
Citação: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Palavras-chave: genômica do SARS-CoV-2, métodos filogenéticos, erros de sequenciamento, variação da taxa de mutação, epidemiologia genômica