Clear Sky Science · pt
Atlas de célula única do córtex cerebral em desenvolvimento na síndrome de Down
Por que esta pesquisa importa
A síndrome de Down é a causa genética mais comum de deficiência intelectual, mas ainda sabemos pouco sobre como ela altera o cérebro humano em desenvolvimento antes do nascimento. Este estudo aprofunda até o nível de células individuais no córtex fetal — a região cerebral crucial para o pensamento e a memória — para mapear o que dá errado, quando isso começa e quais interruptores moleculares podem ser alvo de intervenções.
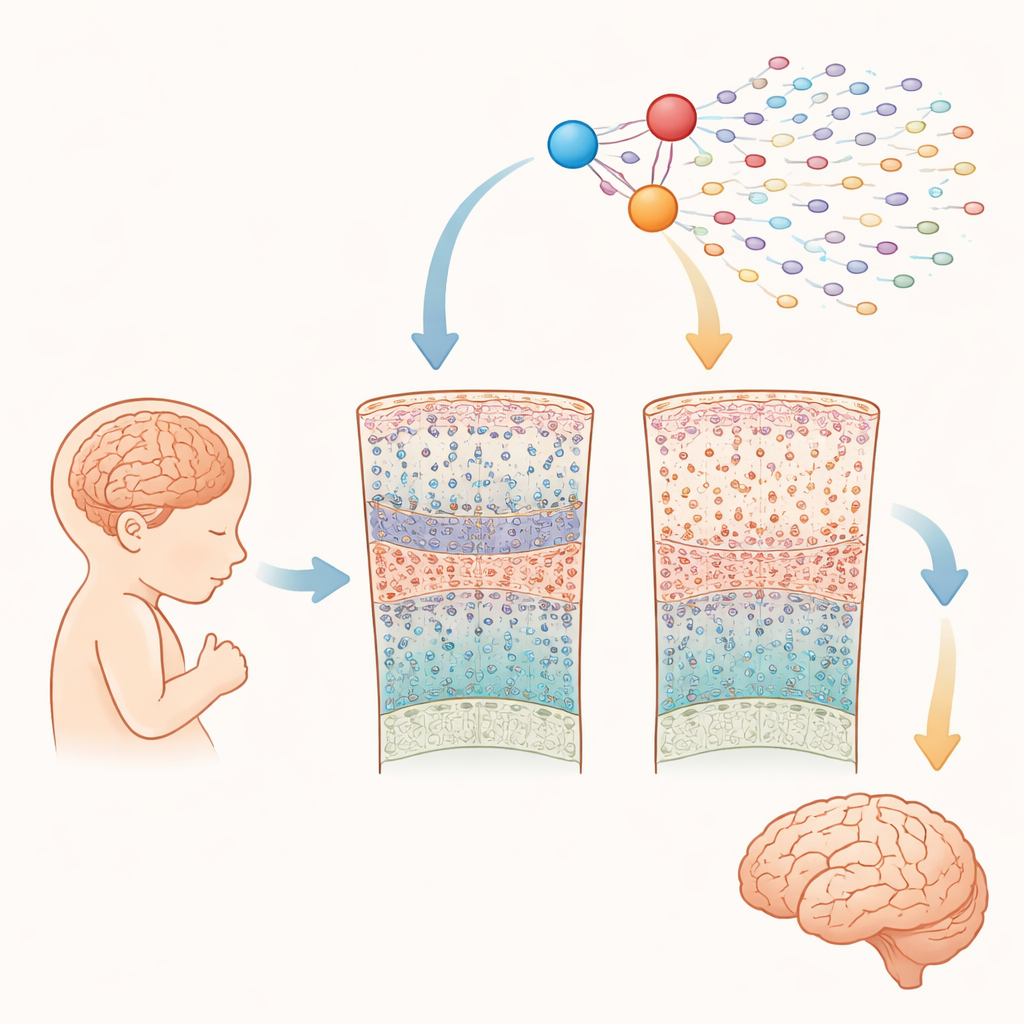
Observando de perto o cérebro em crescimento
Os pesquisadores analisaram quase um quarto de milhão de células extraídas do córtex cerebral de 15 fetos com síndrome de Down e 15 sem, entre 10 e 20 semanas após a concepção. Usando métodos avançados de "célula única", eles mediram tanto quais genes estavam ativados quanto a acessibilidade do DNA em cada célula. Isso permitiu identificar todos os principais tipos celulares presentes nessa fase de gestação média — como células progenitoras com características de células-tronco, diferentes tipos de neurônios excitatórios e inibitórios e células gliais iniciais — e comparar sua abundância e atividade gênica entre cérebros com síndrome de Down e típicos.
Alterações precoces em células-chaves do pensamento
A maioria das classes celulares amplas esteve presente em números semelhantes em ambos os grupos durante essa janela inicial. No entanto, a equipe encontrou uma redução marcante e seletiva de um tipo particular de neurônio excitatório que normalmente ocupa a camada 4 do córtex e é importante para processar informações de entrada. Esses neurônios são definidos por proteínas chamadas RORB e FOXP1. Em fetos com síndrome de Down, os neurônios RORB–FOXP1 já estavam reduzidos na metade da gestação, especialmente entre 16 e 20 semanas, enquanto outros tipos neuronais pareciam relativamente preservados. Isso sugere que problemas na geração ou maturação desse subconjunto de células começam in utero e podem contribuir diretamente para dificuldades cognitivas posteriores.
Programas genéticos interrompidos e interruptores mestres
Além das contagens celulares, o estudo revelou que centenas de genes estavam sutilmente desregulados, particularmente em neurônios excitatórios e seus progenitores. Muitos desses genes estão envolvidos na construção do prosencéfalo, na modelagem dos ramos neuronais, na formação de conexões e no suporte às funções cerebrais superiores. Em vez de apenas as cópias extras de aproximadamente 200 genes no cromossomo 21 atuarem isoladamente, os dados apontam para uma rede perturbada de regulação gênica. Usando uma abordagem que combina atividade gênica com acessibilidade do DNA, os autores mapearam circuitos regulatórios e destacaram três fatores de transcrição — BACH1, PKNOX1 e GABPA — codificados no cromossomo 21 como "hubs" sensíveis à dosagem. Essas moléculas parecem influenciar outros reguladores críticos do desenvolvimento cortical, incluindo fatores já ligados à deficiência intelectual, e ajudam a explicar como um aumento modesto de 1,5 vez na dosagem gênica pode repercutir por programas de desenvolvimento inteiros.
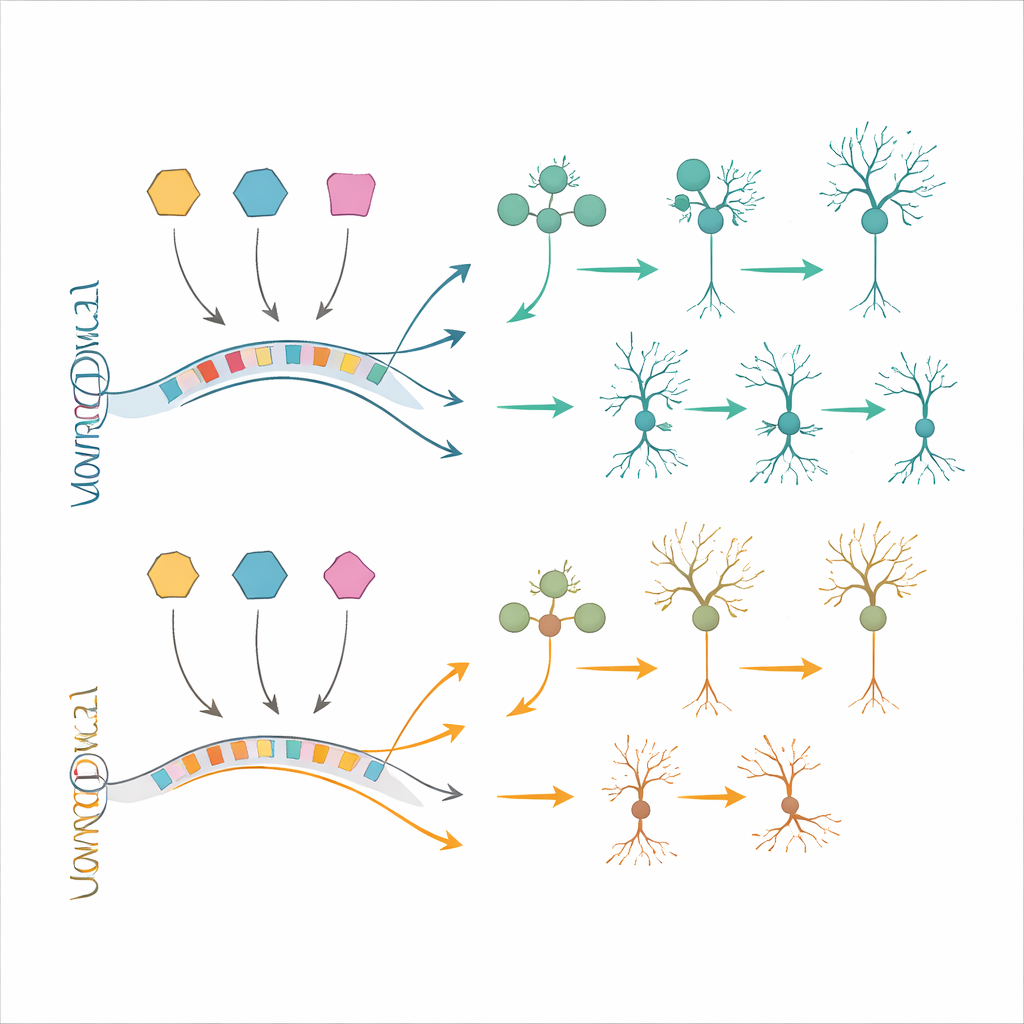
Testando estratégias de resgate em células e cérebros vivos
Para verificar se essas mudanças moleculares poderiam ser corrigidas, a equipe recorreu a modelos de células-tronco. Eles geraram células progenitoras neurais e neurônios a partir de células-tronco pluripotentes induzidas com trissomia 21 e de células controle pareadas. Muitas das alterações de expressão gênica observadas em tecido fetal reapareceram nessas células cultivadas em laboratório, confirmando a relevância dos modelos. Os pesquisadores então usaram oligonucleotídeos antisense — curtas sequências de material semelhante ao DNA projetadas — para reduzir BACH1, PKNOX1 ou GABPA em direção a níveis normais. Essa normalização parcial dos fatores de transcrição superexpressos levou a uma recuperação parcial de vários genes a jusante, incluindo alguns conhecidos por estarem envolvidos em deficiência intelectual e diferenciação neuronal. Em uma abordagem complementar, transplantaram células neurais humanas com trissomia 21 em cérebros de camundongos e deixaram-nas amadurecer in vivo. Esses enxertos reproduziram características adicionais semelhantes à síndrome de Down, como desequilíbrio neurônio–glia alterado e mudanças gênicas que não foram totalmente capturadas em culturas, oferecendo um campo de testes poderoso para futuras terapias.
O que isso significa para o futuro
Em conjunto, o trabalho fornece um atlas detalhado de como a síndrome de Down remodela o panorama genético do córtex em desenvolvimento com resolução de célula única. Para um leitor geral, a mensagem central é que o cromossomo extra não adiciona simplesmente alguns genes desonestos; ele ajusta muitos interruptores moleculares interconectados, levando a faltas precoces e seletivas de certos neurônios relacionados ao pensamento. Ao identificar um pequeno conjunto de fatores de transcrição do cromossomo 21 como atores centrais — e mostrar que seus efeitos podem ser parcialmente revertidos em células humanas — o estudo abre a porta para estratégias mais direcionadas visando melhorar o desenvolvimento e a função cerebral na síndrome de Down.
Citação: Lattke, M., Tan, W.L., Sukumaran, S.K. et al. Single-cell atlas of the developing Down syndrome brain cortex. Nat Med 32, 1061–1072 (2026). https://doi.org/10.1038/s41591-026-04211-1
Palavras-chave: Síndrome de Down, desenvolvimento do cérebro fetal, genômica de célula única, neurônios corticais, fatores de transcrição