Clear Sky Science · pt
Modelos transferíveis de enantioseletividade a partir de dados esparsos
Uma maneira mais inteligente de encontrar o catalisador certo
Químicos frequentemente buscam melhores medicamentos e materiais tentando unir átomos de carbono em arranjos tridimensionais muito específicos. Obter aquele resultado sutil de “mão direita” versus “mão esquerda” — conhecido como enantioseletividade — normalmente exige testar muitos catalisadores metálicos e condições reacionais por tentativa e erro. Este artigo apresenta uma forma de usar quantidades relativamente pequenas de dados experimentais, combinadas com cálculos computacionais rápidos, para prever quais catalisadores à base de níquel irão conferir a desejada quiralidade em uma ampla gama de reações, potencialmente economizando semanas ou meses de trabalho de laboratório.
Por que moléculas quirais são tão difíceis de controlar
Muitos fármacos e produtos naturais existem em formas imagem-especular que podem se comportar de maneira muito diferente no organismo. Catalisadores que favorecem uma imagem sobre a outra são, portanto, extremamente valiosos. Mas projetar tais catalisadores é complicado. A química quântica tradicional pode, em princípio, calcular qual via reacional é preferida, porém erros mínimos de energia se traduzem em grandes equívocos na seletividade prevista, e os cálculos são lentos. Modelos estatísticos mais simples, por outro lado, são rápidos mas frequentemente ignoram a dança detalhada entre o catalisador metálico e as moléculas reagentes, especialmente quando o mecanismo reacional pode mudar sutilmente conforme diferentes parceiros são usados.
Capturando os momentos importantes em uma reação
Os autores preenchem essa lacuna concentrando-se nas etapas mais críticas de uma reação de acoplamento cruzado catalisada por níquel: os passos em que novas ligações carbono–carbono são formadas e o produto final é liberado. Em vez de executar simulações de alto custo, eles usam um método quântico simplificado para gerar estruturas tridimensionais de estados de transição e intermediários-chave em muitas combinações possíveis de catalisadores e substratos. A partir dessas estruturas, extraem centenas de descritores fisicamente significativos, como o quão congestionado é o ambiente do catalisador perto de certos átomos ou quão facilmente os elétrons podem se mover. Esses números são então inseridos em modelos de regressão linear diretos que conectam características estruturais à seletividade medida.
Aprendendo a partir de dados esparsos para orientar novos experimentos
Um feito central do trabalho é aproveitar ao máximo dados esparsos — as combinações limitadas de catalisadores e substratos tipicamente relatadas em um artigo. Em um estudo de caso, a equipe reexamina uma reação com níquel que acopla oxidas de estireno com iodetos arílicos. Eles mostram que descritores extraídos do estado de transição mais relevante superam aqueles obtidos de fragmentos de catalisador simplificados, mesmo que os cálculos subjacentes sejam mais baratos. Com esses modelos em mãos, testam virtualmente muitos mais ligantes em pares de substratos existentes e identificam novas opções de catalisador que aumentam o excesso enantiomérico em exemplos particularmente difíceis, evitando dezenas de experimentos desnecessários.
Transferindo conhecimento entre diferentes reações
A abordagem é poderosa porque pode ser transferida entre reações de níquel diferentes, mas relacionadas. Em um segundo conjunto de estudos, os autores combinam dados de vários tipos de reações com níquel que formam ligações entre átomos de carbono híbridizados sp3 e parceiros como grupos arílicos ou alquenilos, mesmo quando as condições exatas ou os parceiros de acoplamento diferem. Ao construir modelos a partir dos mesmos descritores mecanisticamente significativos, eles prevêem com sucesso a enantioseletividade para novos ligantes, novas combinações de substratos e até uma classe inteiramente nova de reação formadora de ligação carbono–carbono que não foi incluída no conjunto de treino. A análise de quais descritores importam mais também sugere qual etapa do ciclo catalítico realmente determina a quiralidade para cada família de reações.
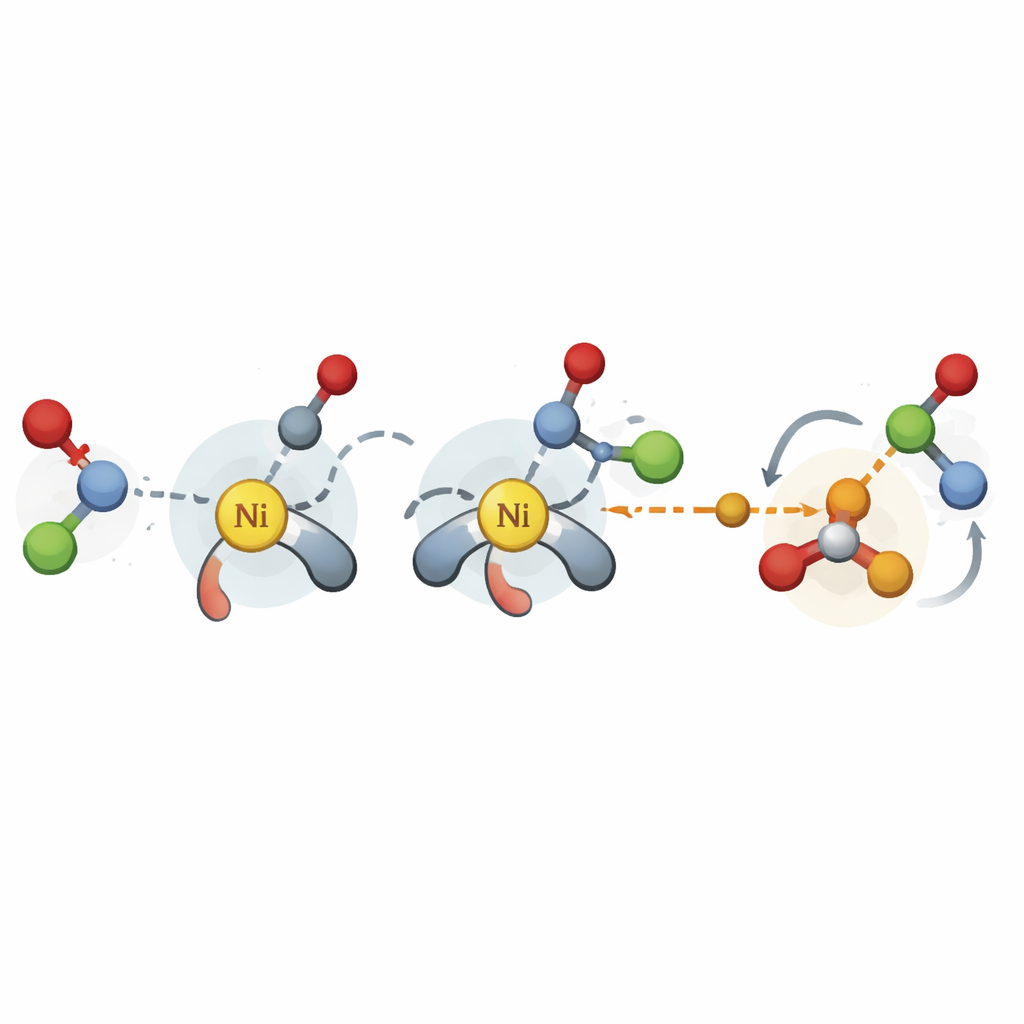
Ajuda para os químicos iniciarem novas reações mais rápido
Em uma demonstração final, os autores usam seu esquema de descritores junto com uma plataforma de otimização bayesiana para projetar um acoplamento catalisado por níquel entre acetais benzílicos e iodetos arílicos que não havia sido desenvolvido assimetricamente antes. Partindo de dados da literatura sobre outras reações, o modelo recomenda pequenos lotes de ligantes promissores para testar, convergindo rapidamente para a classe de melhor desempenho em poucas dezenas de experimentos. Para um químico, isso significa uma ferramenta prática para “start frio” em um novo projeto catalítico: ao inserir um punhado de resultados iniciais, o modelo pode sugerir quais ligantes quirais têm maior probabilidade de fornecer alta enantioseletividade. No conjunto, o estudo mostra que características computacionais baratas e bem escolhidas podem transformar dados passados limitados em orientação amplamente útil para construir a próxima geração de reações seletivas.
Citação: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Palavras-chave: catálise assimétrica, acoplamento cruzado com níquel, aprendizado de máquina em química, otimização de reações, previsão de enantioseletividade