Clear Sky Science · pt
Telas CRISPR bidirecionais decodificam um circuito fibrótico dependente de GLIS3
Quando a cura se transforma em cicatrização prejudicial
Nossos intestinos devem se reparar após cada corte e irritação. Mas em doenças crônicas como a doença de Crohn e a colite ulcerativa, esse processo de cura pode sair dos trilhos, levando à formação de tecido cicatricial espesso e rígido que estreita o intestino e pode demandar cirurgia. Este estudo revela uma conversa oculta entre células imunes e células estruturais do intestino que impulsiona essa cicatrização, e aponta um interruptor mestre, um gene chamado GLIS3, que pode oferecer uma nova forma de romper o ciclo.
Uma rede oculta dentro de intestinos inflamados
Para entender por que alguns pacientes desenvolvem inflamação persistente e fibrose (cicatrização), os pesquisadores criaram um “atlas” celular do intestino humano. Eles combinaram sequenciamento de RNA em célula única, que lê os genes ativos em células individuais, com perfilamento espacial que mapeia onde essas células estão em fatias reais de tecido. Usando amostras de pessoas com doença de Crohn, colite ulcerativa e controles, mapearam mais de quatro milhões de células através da parede intestinal. Dentro dessa multidão, um subgrupo de fibroblastos se destacou: fibroblastos associados à inflamação, ou IAFs. Essas células se aglomeraram em áreas de colite ativa e crônica e exibiram uma assinatura gênica ligada à resistência às terapias padrão anti–TNF, sugerindo que desempenham um papel central em formas difíceis de tratar da doença. 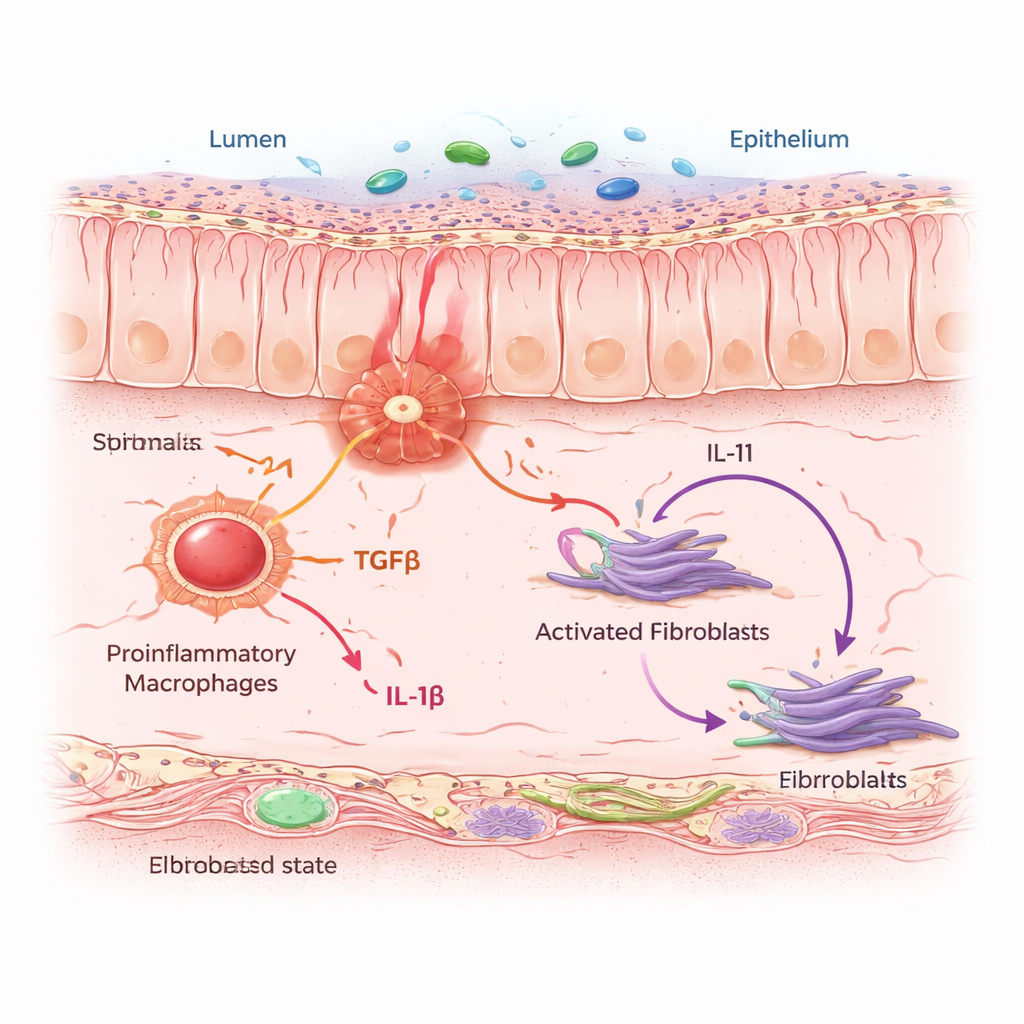
Macrófagos sussurram, fibroblastos cicatrizam
Os IAFs não atuaram sozinhos. Eles se agruparam em “vizinhanças” densas em macrófagos pró-inflamatórios — células imunes que detectam perigo e liberam sinais de alarme. Usando modelos computacionais e experimentos de co-cultura celular, a equipe mostrou que quando os macrófagos são conduzidos a um estado inflamatório, eles secretam duas proteínas mensageiras-chave: TGFβ e IL-1β. Fibroblastos vizinhos captam esses sinais por meio de receptores específicos. Quando ambos os sinais chegam juntos, os fibroblastos mudam para o estado IAF e começam a produzir IL-11, uma citocina já suspeita de promover fibrose, além de colágeno e outras proteínas da matriz que espessam e enrijecem a parede intestinal. Em camundongos submetidos a um regime de colite crônica, bloquear a IL-11 ou deletá-la seletivamente nos fibroblastos reduziu o acúmulo de colágeno sem impedir a inflamação inicial, mostrando que a IL-11 é um motor crucial da fase de cicatrização.
GLIS3: o interruptor mestre nos fibroblastos fibróticos
Para avançar de correlações para mecanismos, os autores usaram poderosas ferramentas CRISPR em escala genômica. Eles modificaram fibroblastos humanos de modo que a produção de IL-11 pudesse ser monitorada por uma etiqueta fluorescente, e então realizaram telas paralelas que ou nocauteavam genes ou os ativavam em todo o genoma. Ao classificar as células que produziam quantidades incomuns de IL-11 após estimulação com TGFβ e IL-1β, identificaram genes que controlam essa resposta. Entre muitos componentes de sinalização, um fator de transcrição — GLIS3 — emergiu como um regulador principal. Quando GLIS3 foi desativado, os fibroblastos produziram muito menos IL-11; quando foi aumentado, a IL-11 disparou. Experimentos adicionais mostraram que GLIS3 se desloca para o núcleo do fibroblasto em resposta aos sinais dos macrófagos, liga-se diretamente ao DNA próximo ao gene IL11 e a outros, e ativa um amplo programa de genes inflamatórios e fibróticos, incluindo colágenos e fatores que atraem mais células imunes. 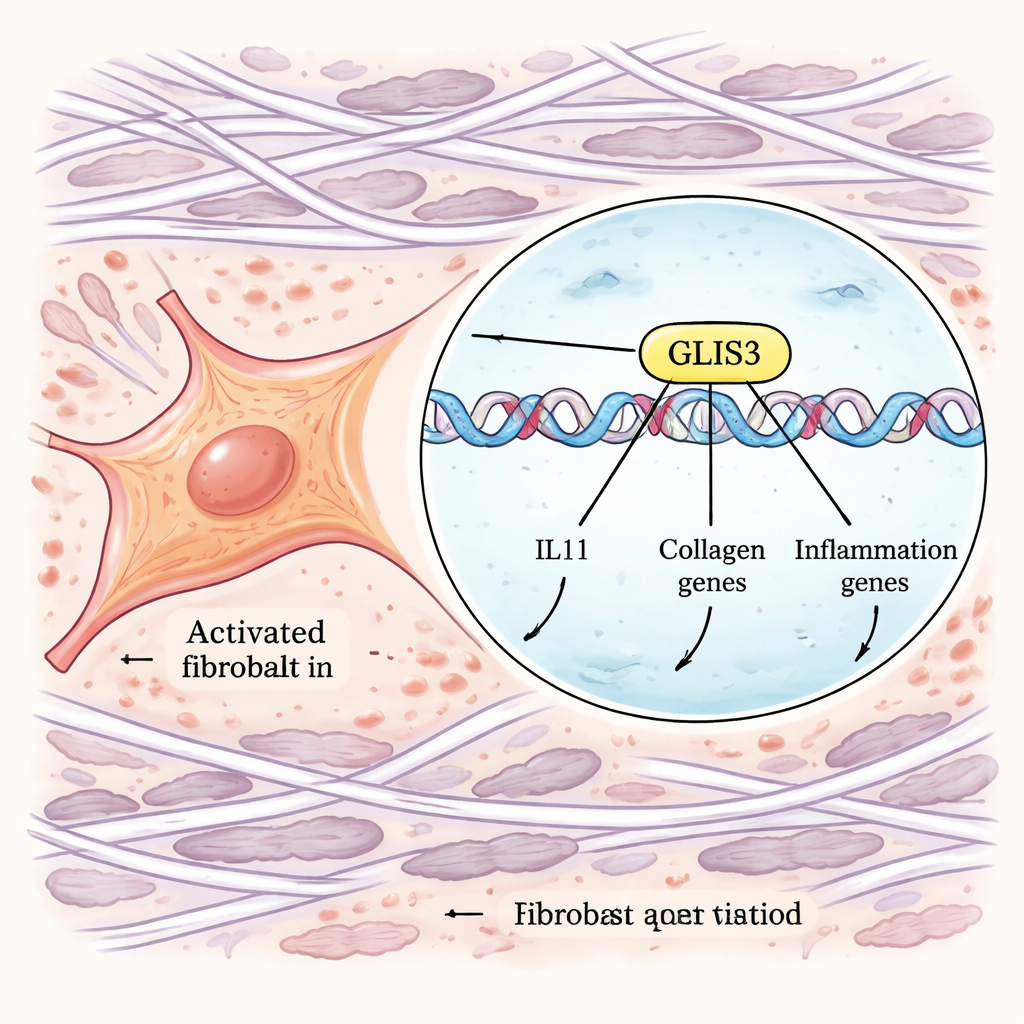
De modelos em camundongos à gravidade em pacientes
A equipe então perguntou se esse programa dirigido por GLIS3 tem importância em organismos vivos. Em camundongos, eles criaram uma linhagem na qual GLIS3 podia ser removido apenas dos fibroblastos. Quando esses animais foram submetidos à colite crônica, desenvolveram menos cicatrizes intestinais, apresentaram níveis mais baixos de colágeno e expressão de genes fibróticos, e mostraram inflamação reduzida em comparação com camundongos normais. O mapeamento espacial confirmou que os camundongos deficientes em GLIS3 tinham menos fibroblastos produtores de IL-11 e menos macrófagos e neutrófilos ativados nas proximidades, indicando que interromper o GLIS3 enfraquece todo o circuito inflamatório-fibrótico. Ao analisar uma grande coorte pediátrica de colite ulcerativa, os autores extraíram uma “assinatura” de GLIS3 de 50 genes e descobriram que sua atividade em biópsias do cólon acompanhava de perto a gravidade da doença e a abundância de IAFs e macrófagos ativados, ligando essa via diretamente aos desfechos dos pacientes.
Quebrando o ciclo de inflamação e cicatrização
Para não especialistas, a conclusão é que este trabalho revela um circuito auto-reforçador: macrófagos inflamatórios acionam fibroblastos para se tornarem IAFs formadores de cicatriz; esses IAFs, sob controle do GLIS3, produzem IL-11, colágeno e outros fatores que remodelam o tecido e atraem mais células inflamatórias. Medicamentos padrão que suprimem amplamente o sistema imunológico podem não romper completamente esse ciclo, o que ajuda a explicar por que muitos pacientes eventualmente falham nas terapias existentes. Ao identificar GLIS3 e o estado de fibroblasto produtor de IL-11 como nós centrais no circuito inflamação–fibrose, este estudo aponta para estratégias mais direcionadas — voltadas aos fibroblastos, e não apenas às células imunes — que poderiam um dia prevenir ou reverter a cicatrização na doença inflamatória intestinal e possivelmente em outras condições inflamatórias crônicas.
Citação: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Palavras-chave: doença inflamatória intestinal, fibrose intestinal, fibroblastos, macrófagos, GLIS3