Clear Sky Science · pt
O papel crítico de defeitos intrínsecos e interações de muitos corpos na estabilidade do MnBi2Te4
Por que pequenas falhas em cristais importam para a tecnologia do futuro
Muitas das tecnologias quânticas de amanhã — como eletrônicos ultraeficientes e novos tipos de computadores — dependem de materiais exóticos cujas superfícies conduzem eletricidade enquanto seus interiores permanecem isolantes. Um dos mais promissores é o MnBi2Te4, um “ímã topológico” que pode abrigar correntes de borda sem resistência úteis para dispositivos de baixo consumo e computação quântica. Mas, em cristais reais, átomos frequentemente ocupam posições incorretas, e essas pequenas falhas podem silenciosamente destruir os efeitos que os engenheiros desejam explorar. Este estudo faz uma pergunta básica, porém crucial: essas falhas são um acidente de fabricação ou são, na verdade, favorecidas pela natureza nas temperaturas em que o material é produzido?
Um material promissor com um problema persistente
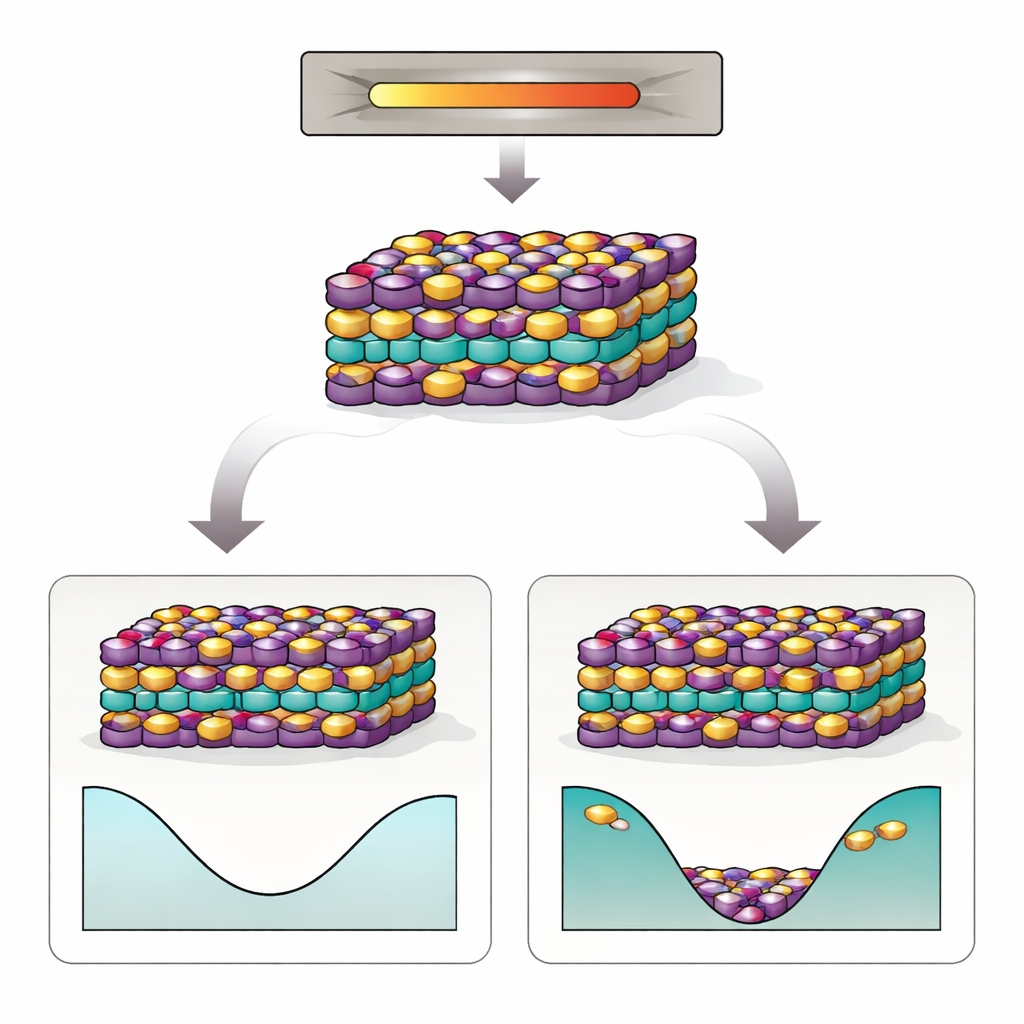
MnBi2Te4 é formado por folhas atômicas empilhadas, como um sanduíche cuidadosamente ordenado. Seu comportamento eletrônico especial depende de duas coisas: um arranjo preciso dos átomos de manganês (Mn), bismuto (Bi) e telúrio (Te), e um padrão delicado de alinhamento magnético entre as camadas. No entanto, experimentos repetidamente encontram muitos átomos de Mn e Bi trocando de lugar — os chamados defeitos antisítios. Essas trocas embaralham o padrão magnético, afastam o material do seu estado isolante ideal e tornam mais difícil observar os fenômenos quânticos desejados. Pior ainda, mesmo quando os cristais são crescidos e recozidos com muito cuidado, os defeitos antisítios permanecem teimosamente, sugerindo que algo mais profundo que o processamento imperfeito está em ação.
Por que cálculos anteriores discordavam dos experimentos
Simulações computacionais padrão apresentavam um quadro puzzling. No zero absoluto, métodos quântico‑mecânicos comuns previam que criar uma troca Mn–Bi custa energia e, portanto, deveria ser raro. Isso entra em conflito com experimentos que mostram altos níveis de defeitos em amostras reais produzidas em torno de 850 kelvin (mais de 500 °C). Os autores argumentam que duas peças-chave estavam faltando na teoria anterior. Primeiro, os defeitos eram geralmente tratados um a um, ignorando como eles interagem e se agrupam. Segundo, os cálculos eram tipicamente feitos a temperatura zero, negligenciando como o calor e a desordem alteram quais arranjos atômicos são favorecidos. Em um material que já é apenas marginalmente estável, mesmo pequenas contribuições do comportamento de “muitos corpos” dos elétrons e do grande número de arranjos possíveis podem alterar o equilíbrio.
Acompanhando cada troca em um cristal virtual
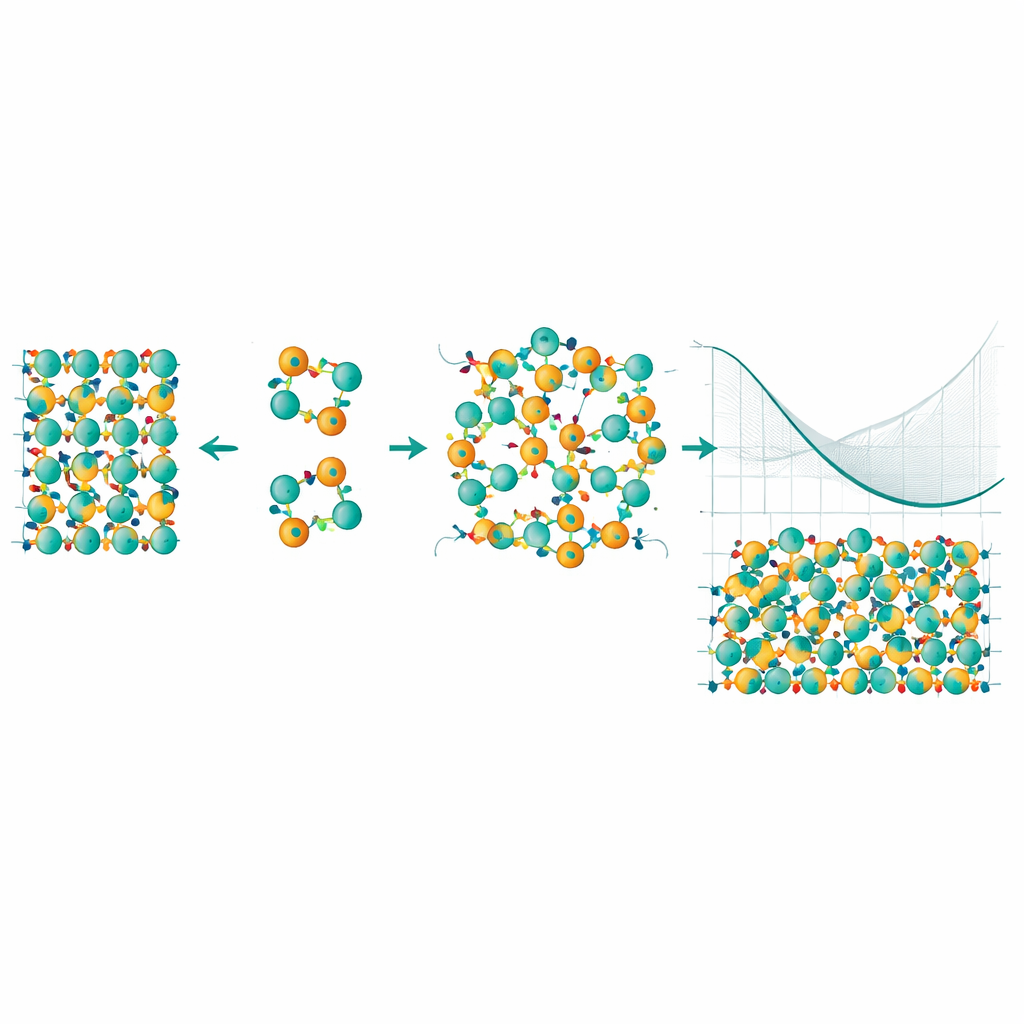
Para enfrentar isso, os pesquisadores construíram um modelo estatístico capaz de explorar milhões de maneiras diferentes pelas quais os átomos de Mn e Bi podem se reorganizar. Eles usaram uma técnica chamada expansão em clusters, que decompõe a energia do cristal em contribuições de átomos individuais, pares e pequenos grupos, e então combinaram isso com amostragem de Monte Carlo para ver quais padrões aparecem a diferentes temperaturas. De modo crucial, corrigiram as energias subjacentes usando um método especialmente preciso conhecido como Monte Carlo quântico, que captura melhor as sutis interações elétron‑elétron. Essa abordagem híbrida permitiu calcular não apenas o custo energético de uma única troca, mas como esse custo muda à medida que mais defeitos aparecem e começam a influenciar uns aos outros.
Quando a desordem se torna a opção mais barata
As simulações revelam que as interações entre múltiplos defeitos antisítios e a “entropia configuracional” da desordem — essencialmente o enorme número de maneiras de arranjar os átomos trocados — remodelam dramaticamente o comportamento do material nas temperaturas de crescimento. Embora uma troca isolada Mn–Bi seja custosa a temperatura zero, em temperaturas mais altas o ganho em entropia compensa esse custo energético. Os autores identificam uma transição ordem‑desordem perto da temperatura de síntese: acima desse ponto, átomos Mn e Bi trocados tornam‑se termodinamicamente favorecidos, e a energia livre de um cristal defeituoso cai abaixo da de um cristal perfeitamente ordenado. Em outras palavras, a natureza prefere um cristal com uma fração substancial de defeitos antisítios, e esses defeitos tendem a se formar em aglomerados correlacionados em vez de aparecer aleatoriamente.
O que isso significa para fabricar materiais quânticos melhores
Para não especialistas, a conclusão principal é que os defeitos problemáticos em MnBi2Te4 não são simplesmente uma falha de fabricação; eles são uma consequência natural da termodinâmica do material nas temperaturas em que é crescido. O estudo mostra que, uma vez incluídas corretamente as interações de muitos corpos e a estatística da desordem, teoria e experimento finalmente concordam: defeitos antisítios se formam espontaneamente e em grande número. Esse insight explica por que produzir cristais verdadeiramente livres de defeitos tem sido tão difícil e oferece um roteiro para melhorar outros materiais quânticos delicados. Qualquer esforço para projetar amostras melhores — ajustando condições de crescimento, composições ou rotas de processamento — precisa enfrentar o fato de que, em alta temperatura, a desordem não é um acidente, mas a escolha de menor energia para o cristal.
Citação: Ghaffar, A., Saritas, K. & Reboredo, F.A. The critical role of intrinsic defects and many-body interactions on the stability of MnBi2Te4. npj Comput Mater 12, 119 (2026). https://doi.org/10.1038/s41524-026-02019-8
Palavras-chave: isolantes topológicos, materiais magnéticos, defeitos cristalinos, Monte Carlo quântico, termodinâmica de materiais