Clear Sky Science · pt
Potenciais de aprendizagem ativa para diagramas de fases de primeiros princípios usando amostragem aninhada com troca de réplicas
Por que isso importa para materiais do futuro
De chips de computador mais rápidos a peças de avião mais resistentes, muitas tecnologias modernas dependem de saber como um material muda quando é aquecido ou submetido a pressão. Essas mudanças, chamadas transições de fase, são resumidas em diagramas de fase — os mapas que informam aos cientistas qual forma de um material é estável em certas condições. Este estudo apresenta uma nova forma de traçar automaticamente esses mapas diretamente a partir de cálculos quântico‑mecânicos, usando inteligência artificial para reduzir drasticamente o custo mantendo alta precisão.
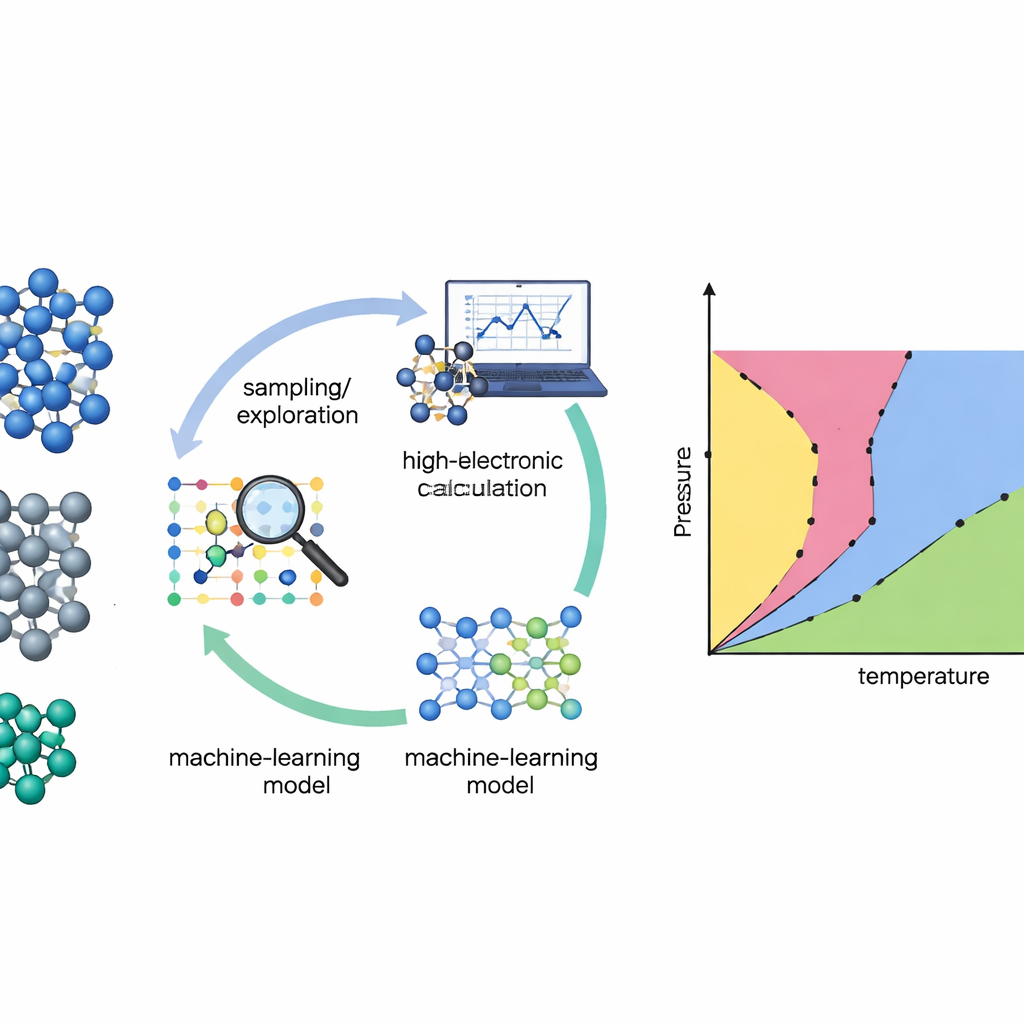
Mapeando materiais sem suposições
Tradicionalmente, construir um diagrama de fase a partir de primeiros princípios é como caminhar por uma paisagem acidentada no escuro: é preciso já suspeitar onde estão os vales e os passes importantes. Muitos métodos padrão funcionam apenas se os pesquisadores fornecerem conhecimento prévio forte sobre quais estruturas cristalinas ou “caminhos” explorar. Os autores, em vez disso, recorrem a uma técnica chamada amostragem aninhada, que vasculha sistematicamente toda a paisagem energética de um material sem assumir quais fases surgirão. Ao acompanhar quão acessíveis são diferentes regiões dessa paisagem, a amostragem aninhada pode recuperar propriedades termodinâmicas e mudanças de fase em uma ampla faixa de temperaturas em uma única varredura.
Deixando o modelo escolher o que precisa aprender
Mesmo o método de busca mais inteligente precisa de uma boa descrição de como os átomos interagem. Cálculos quântico‑mecânicos diretos (teoria do funcional da densidade) são precisos, mas caros demais para serem avaliados milhões ou bilhões de vezes. A equipe resolve isso treinando potenciais interatômicos por aprendizado de máquina — modelos rápidos que imitam as forças quânticas entre átomos. O problema é que tais modelos são confiáveis apenas onde viram exemplos suficientes. Para contornar isso, os autores constroem um ciclo de aprendizagem ativa: o modelo de aprendizado de máquina executa a simulação de amostragem aninhada, identifica configurações nas quais está incerto e então solicita cálculos quântico‑mecânicos de alto nível apenas nesse subconjunto cuidadosamente escolhido. Os novos dados são incorporados ao modelo, que se torna mais confiável nas regiões que mais importam para o diagrama de fases.
Um novo motor para explorar silício, germânio e titânio
Os pesquisadores testaram a abordagem em três elementos importantes: silício e germânio, semicondutores bem conhecidos, e titânio, um metal estrutural amplamente usado. Começaram com bancos de dados iniciais modestos construídos a partir de estruturas cristalinas conhecidas e pequenas distorções, omitindo deliberadamente líquidos e muitas configurações de alta energia. A amostragem aninhada com troca de réplicas — muitas execuções de amostragem aninhada a diferentes pressões que podem trocar configurações — então explorou as paisagens energéticas dos materiais. Após cada rodada de exploração, o algoritmo selecionou automaticamente centenas de configurações atômicas representativas, com peso para aquelas em que as previsões de força mais divergiam entre um comitê de modelos de rede neural. Essas foram recalculadas com um método quântico de alta precisão (r2SCAN) e usadas para retreinar os potenciais antes de iniciar a próxima rodada.
De começos ruidosos a mapas de fase confiáveis
Em cerca de dez a quinze ciclos de aprendizagem, a incerteza dos modelos diminuiu de forma constante, especialmente nas forças que governam o movimento atômico. Ao mesmo tempo, as trajetórias de amostragem aninhada começaram a revelar os contornos familiares dos diagramas de fase. Para o silício, o método reproduziu a conhecida estrutura diamantada em baixa pressão, sua fase hexagonal em alta pressão e o comportamento característico de fusão com temperatura e pressão, tudo em bom acordo com experimentos e simulações anteriores. O germânio mostrou um padrão semelhante, com uma fase tipo diamante em baixa pressão cedendo lugar a uma fase metálica em alta pressão, embora a pressão exata de transição tenha se deslocado um pouco por causa da aproximação quântico‑mecânica escolhida. O titânio ofereceu um teste mais difícil: suas fases são metálicas, estruturalmente similares e separadas por pequenas diferenças de energia. Mesmo assim, a estratégia de aprendizagem ativa capturou a sequência de fases sólidas e a linha de fusão, e verificações adicionais usando funções de distribuição radial confirmaram as identidades das estruturas previstas.
O que isso significa para projetar novos materiais
Em termos simples, o estudo mostra que um computador agora pode aprender por si só como um material se comporta numa ampla faixa de temperaturas e pressões, consultando um “oráculo” quântico‑mecânico apenas quando necessário. O motor de amostragem aninhada com troca de réplicas garante exploração ampla e imparcial, enquanto o ciclo de aprendizagem ativa assegura que os potenciais de aprendizado de máquina sejam precisos onde isso tem importância termodinâmica. Embora o trabalho atual se concentre em três elementos e num método quântico particular, a estrutura é geral: pode ser pareada com teorias eletrônicas mais avançadas ou redes neurais mais potentes, e estendida a ligas ou compostos complexos. À medida que a capacidade computacional e os algoritmos avançam, esse tipo de fluxo de trabalho autônomo pode tornar‑se uma ferramenta padrão para prever diagramas de fase e orientar a descoberta de novos materiais com propriedades sob medida.
Citação: Unglert, N., Ketter, M. & Madsen, G.K.H. Active learning potentials for first-principles phase diagrams using replica-exchange nested sampling. npj Comput Mater 12, 107 (2026). https://doi.org/10.1038/s41524-026-01989-z
Palavras-chave: diagramas de fase de materiais, aprendizagem ativa, potenciais por aprendizado de máquina, amostragem aninhada, silício germânio titânio