Clear Sky Science · pt
Mistura espacial eficiente e precisa de potenciais interatômicos aprendidos por máquina para ciência dos materiais
Por que simulações atômicas mais rápidas importam
Projetar materiais melhores para tecnologias como fusão nuclear, microeletrônica e ligas estruturais depende cada vez mais de simulações por computador que acompanham como os átomos se movem e interagem. Os métodos mais precisos emprestam conceitos da física quântica, mas são tão exigentes computacionalmente que apenas tamanhos de sistema e escalas de tempo modestos são práticos. Este artigo apresenta o ML‑MIX, uma técnica e pacote de software que permite aos pesquisadores manter a precisão quase quântica exatamente onde ela é necessária, ao mesmo tempo em que usa modelos mais simples e baratos no restante. O resultado é um aumento substancial de velocidade — frequentemente um fator de 4 a 10 — sem perder confiabilidade nas previsões físicas essenciais.
Mesclando visões detalhadas e simples dos átomos
No cerne do trabalho está uma ideia simples: nem todo átomo em uma simulação precisa do mesmo nível de atenção. Regiões onde ligações estão se esticando, rompendo ou se rearranjando — como defeitos, superfícies ou partículas implantadas — beneficiam‑se de potenciais interatômicos modernos baseados em aprendizado de máquina, que imitam a precisão quântico‑mecânica. Mas átomos distantes desses “pontos quentes” em sua maioria vibram em torno de posições regulares e podem ser descritos por modelos muito mais simples. O ML‑MIX fornece uma maneira de combinar um modelo preciso, porém caro, com outro mais enxuto e “barato” dentro da mesma caixa de simulação. Faz isso definindo uma zona central que usa o modelo caro, um buffer circundante onde as forças são cuidadosamente misturadas entre os modelos, e uma zona de volume externo que usa apenas a descrição barata. 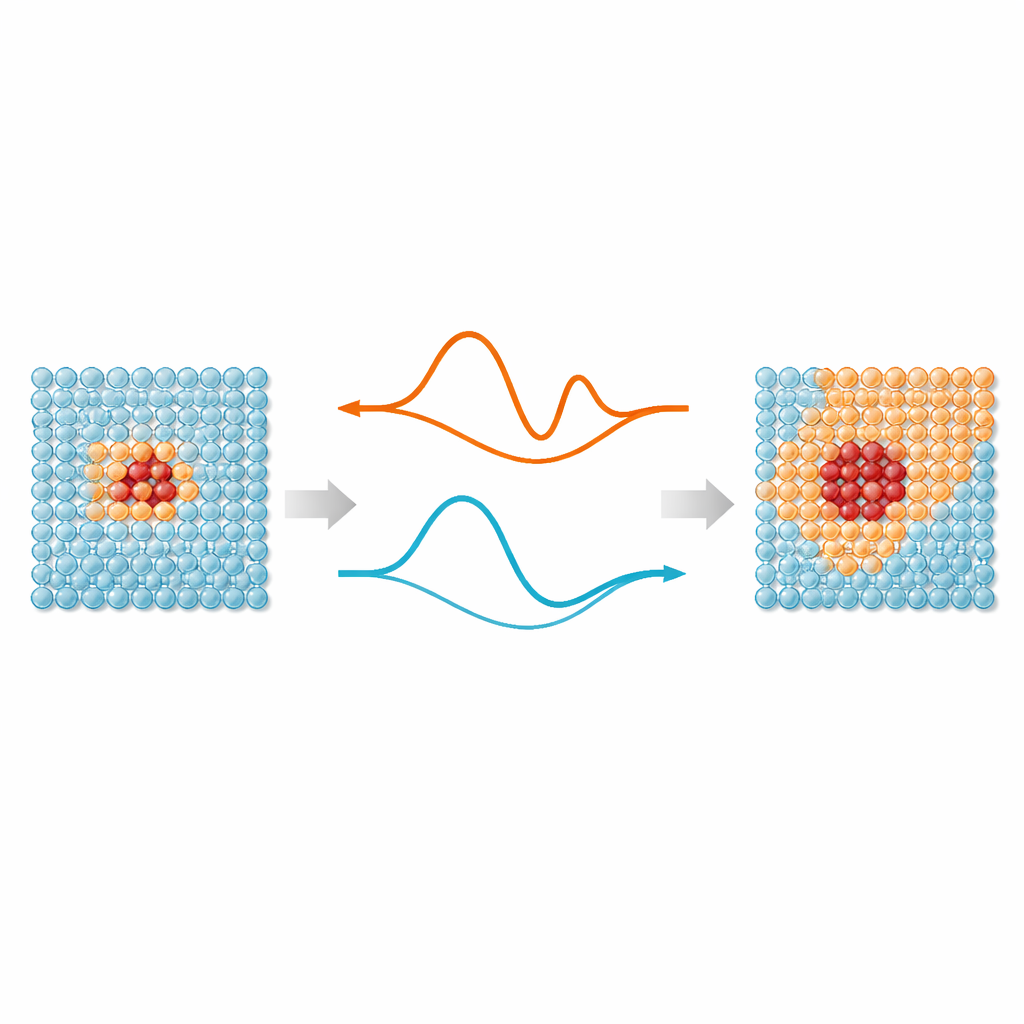
Treinando um modelo barato para imitar um preciso
Um desafio fundamental é garantir que o modelo barato se comporte como o preciso onde eles se encontram. Em vez de ajustar o modelo barato diretamente a um vasto e variado conjunto de dados quântico‑mecânicos, os autores geram dados “sintéticos” focalizados executando o modelo preciso nas condições específicas relevantes para a região de volume: vibrações em alta temperatura e cristais levemente deformados. Em seguida, ajustam o modelo barato para que corresponda a esses dados, impondo restrições rígidas em propriedades materiais básicas, como as constantes elásticas e o espaçamento da rede. Esse ajuste com restrições assegura que tensões e deformações de longo alcance casem suavemente através da fronteira entre os dois modelos, evitando forças artificiais que poderiam corromper a dinâmica perto da interface.
Testando o método
Para verificar se o ML‑MIX realmente funciona, os autores executam uma bateria de testes em sistemas de silício, ferro e tungstênio. Como exemplo simples, eles calculam a barreira de energia para uma vacância — um sítio de rede vazio — no silício mover‑se de uma posição para outra. A simulação mista reproduz o resultado de um cálculo totalmente caro com precisão de cerca de um milésimo de elétron‑volt, enquanto roda cerca de cinco vezes mais rápido. Em um cenário mais dinâmico, eles esticam uma única ligação de silício em um cristal quente e medem a força média sobre ela. Uma simulação que usa apenas o modelo barato já chega surpreendentemente perto, mas quando se adiciona um pequeno núcleo caro ao redor da ligação esticada, o acordo torna‑se estatisticamente indistinguível da referência totalmente precisa, com ganhos de velocidade de até cerca de 13 vezes em execuções seriais.
Acompanhando defeitos e partículas em movimento
Testes mais realistas examinam como defeitos se movem através de metais. A equipe simula a difusão de um defeito auto‑intersticial no ferro e de átomos de hélio dentro do tungstênio. Em cada caso, o modelo caro é confinado a uma pequena região móvel ao redor do defeito, enquanto o restante do cristal é tratado pelo potencial barato. Os coeficientes de difusão resultantes coincidem com os de simulações totalmente precisas dentro do erro estatístico, mesmo quando uma simulação somente com o modelo barato falharia. Os autores então aplicam o método a problemas maiores, cientificamente importantes, no tungstênio, um material candidato para reatores de fusão. Eles modelam o movimento de discordâncias parafuso — defeitos em forma de linha que controlam a deformação plástica — e a implantação de átomos de hélio em uma superfície de tungstênio quente. Em ambos os casos, o ML‑MIX reproduz os resultados obtidos apenas com o modelo caro enquanto reduz o custo computacional por fatores de cerca de quatro a onze. 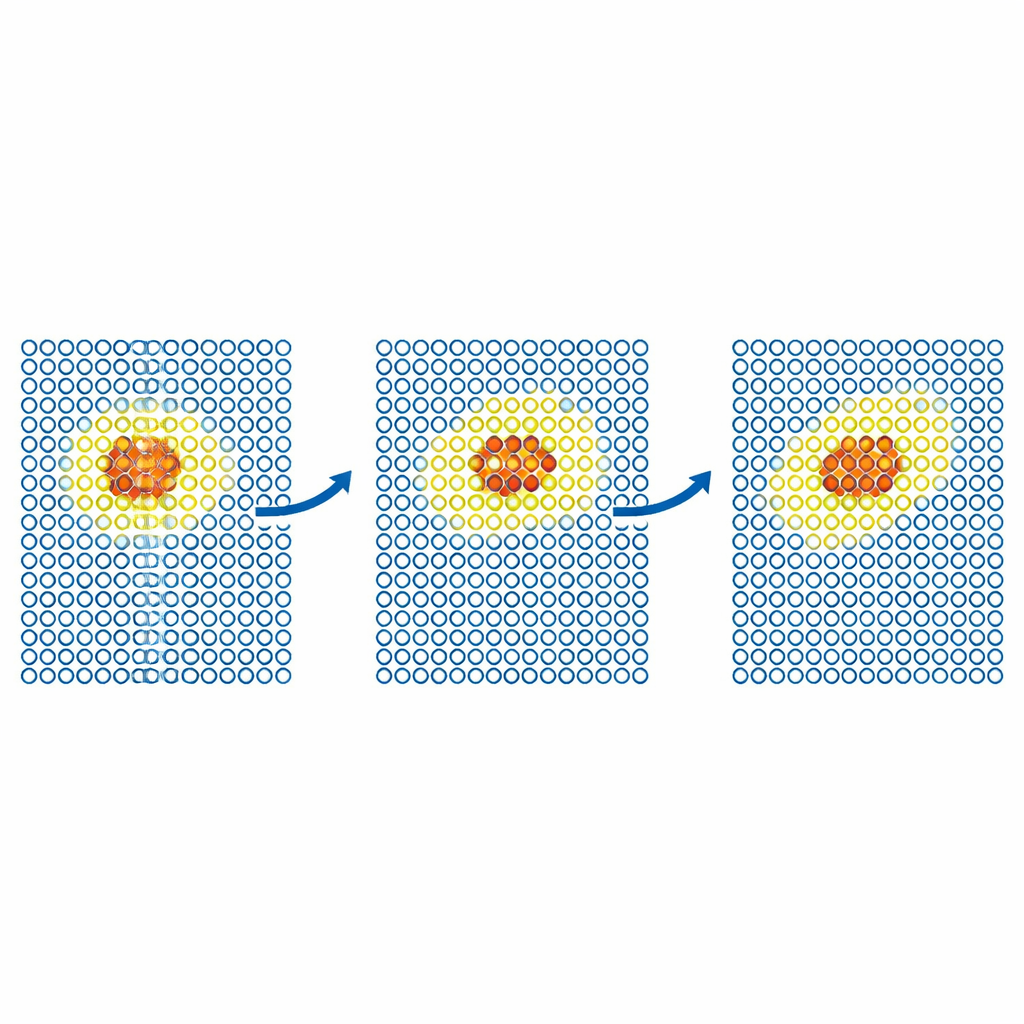
Conferindo com experimentos e perspectivas
O estudo de implantação de hélio mostra com clareza o poder dessa abordagem. Ao misturar um modelo de ponta baseado em aprendizado de máquina para interações hélio–tungstênio com um potencial mais rápido para tungstênio puro, os autores simulam muito mais eventos de impacto e amostras maiores do que seriam viáveis de outra forma, tudo em processadores gráficos. A fração prevista de átomos de hélio que ricocheteiam na superfície versus que se implantam no metal concorda com medições experimentais até energias de incidência de cerca de 80 elétron‑volts, algo que simulações anteriores tinham dificuldade em alcançar. Embora o esquema de mistura não conserve estritamente a energia e requeira termostatos suaves, a deriva resultante é pequena e administrável. De modo geral, o ML‑MIX demonstra que combinar cuidadosamente modelos atômicos detalhados e simplificados pode romper barreiras de longa data entre precisão e escala, abrindo caminho para simulações rotineiras de alta fidelidade de materiais complexos em ambientes realistas.
Citação: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Palavras-chave: potenciais interatômicos aprendidos por máquina, simulação multiescala de materiais, implantação de hélio em tungstênio, defeitos e discordâncias, aceleração de dinâmica molecular