Clear Sky Science · pt
Fluxo de trabalho automatizado assistido por potencial de aprendizado de máquina auto-otimizante para design de materiais de sistemas complexos altamente eficientes
Buscas mais inteligentes por novos materiais
Projetar novos materiais é um pouco como procurar uma agulha em um palheiro quase infinito. De baterias melhores e computadores mais rápidos a lasers mais eficientes e possíveis supercondutores à temperatura ambiente, muitas tecnologias futuras dependem de descobrir arranjos atômicos específicos. Este artigo apresenta uma forma de permitir que a inteligência artificial faça a maior parte dessa busca automaticamente, reduzindo drasticamente o tempo e o custo necessários para encontrar compostos promissores.
Por que o quebra-cabeça dos materiais é tão difícil
As propriedades de um sólido — quão bem conduz eletricidade, quão resistente é, como responde à luz — são determinadas por como seus átomos se organizam em padrões tridimensionais chamados estruturas cristalinas. Em teoria, pode-se usar a mecânica quântica para calcular quais arranjos são estáveis e quais serão suas propriedades. Na prática, esses cálculos quânticos são tão exigentes que apenas uma fração ínfima de todos os materiais possíveis pode ser verificada. O desafio cresce rapidamente quando há mais de dois elementos químicos envolvidos, porque o número de combinações e arranjos atômicos explode, tornando uma busca cega inviável.
Deixar um modelo de aprendizado substituir a física quântica
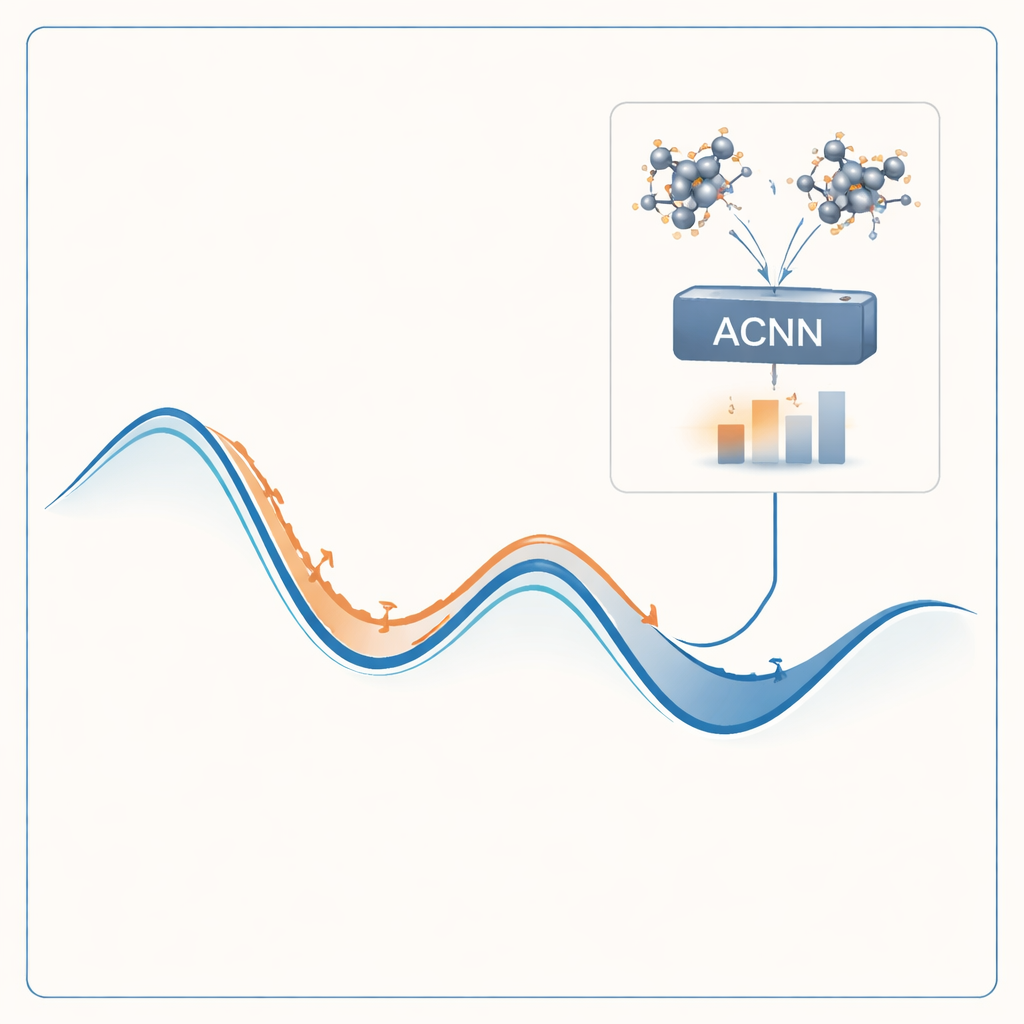
Para enfrentar esse problema, os autores constroem um modelo de aprendizado de máquina capaz de imitar os resultados de cálculos quânticos caros a uma fração do custo. Seu modelo, chamado rede neural acoplada por atenção (ACNN), aprende como a energia de um material depende das posições e dos tipos de seus átomos. Uma vez treinado, ele pode estimar muito rapidamente se uma estrutura cristalina proposta tem probabilidade de ser estável e quais forças atuam em cada átomo. De forma crucial, o modelo é projetado para respeitar requisitos físicos básicos, como o fato de que deslocar ou rotacionar todo o cristal não deve alterar sua energia total.
Um ciclo de descoberta de materiais que se autoaperfeiçoa
Em vez de treinar o modelo uma única vez e torcer para que funcione em toda parte, os autores o envolvem em um ciclo auto-otimizante. O processo começa com um pequeno conjunto de estruturas cristalinas aleatórias, que são avaliadas com cálculos quânticos completos e usadas para treinar uma ACNN inicial. Esse modelo é então usado para relaxar milhões de estruturas de teste, encontrando rapidamente mínimos locais de energia — candidatos a fases estáveis ou quase estáveis. O fluxo de trabalho sinaliza automaticamente dois tipos de estruturas especialmente valiosas: aquelas que parecem muito estáveis e aquelas que aparecem como não físicas ou suspeitas. Somente esses casos selecionados são enviados de volta ao solucionador quântico caro, e os novos resultados são incorporados ao modelo para re-treinamento. Ao longo de muitas rodadas, o modelo fica progressivamente mais preciso nas regiões do espaço de estruturas que importam mais.
Colocando o método à prova
A equipe demonstra sua abordagem em dois sistemas exigentes. O primeiro é uma mistura de alta pressão de magnésio, cálcio e hidrogênio, uma família de compostos de grande interesse para supercondutividade em altas temperaturas. Ao explorar quase seis milhões de estruturas de teste, seu fluxo de trabalho descobriu uma nova fase estável, MgCa₃H₂₃, e várias estruturas “gaiola” ricas em hidrogênio intimamente relacionadas. Cálculos sugerem que algumas dessas poderiam se tornar supercondutoras a temperaturas acima do ponto de ebulição do nitrogênio líquido sob pressão extrema. O segundo teste mira um sistema de quatro elementos contendo berílio, fósforo, nitrogênio e oxigênio, escolhido por seu potencial para abrigar cristais que convertem eficientemente luz de laser em comprimentos de onda no ultravioleta profundo. Nesse caso, o método relaxou mais de nove milhões de estruturas e identificou três fases termodinamicamente estáveis com bandas de gap muito largas e características ópticas promissoras.
Da força bruta à descoberta guiada
Em ambos os exemplos, o fluxo de trabalho automatizado alcança acelerações da ordem de dez mil vezes em comparação com o uso exclusivo de cálculos quânticos, ao mesmo tempo em que continua a identificar de forma confiável estruturas que merecem estudo aprofundado. Para um não especialista, a mensagem-chave é que grande parte da descoberta de materiais agora pode ser tratada por um sistema de aprendizado que aprende onde está incerto e solicita cálculos direcionados de alta precisão apenas quando necessário. Esse tipo de busca autocorretiva assistida por IA abre a porta para explorar misturas de elementos muito mais complexas do que era anteriormente viável, aumentando as chances de descobrir novos supercondutores, cristais ópticos e outros materiais funcionais que sustentam tecnologias de próxima geração.
Citação: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Palavras-chave: descoberta de materiais, potenciais de aprendizado de máquina, predição de estrutura cristalina, hidretos supercondutores, cristais ópticos não lineares