Clear Sky Science · pt
Cálculo de primeira-princípios de estruturas de discordância e transformações de fase induzidas por tensão em óxidos em camadas para baterias íon-sódio
Por que defeitos minúsculos importam para as baterias do futuro
À medida que o mundo olha além do lítio em direção a baterias íon-sódio mais baratas e abundantes, um mundo oculto dentro dos materiais do cátodo torna-se crucial: defeitos cristalinos minúsculos chamados discordâncias. Essas irregularidades lineares, da largura de poucos átomos, ajudam o material a se deformar à medida que íons de sódio entram e saem — mas também podem desencadear danos estruturais que encurtam a vida útil da bateria. Este artigo usa simulações computacionais em nível quântico para revelar como as discordâncias se formam, se movem e impulsionam mudanças de fase em cátodos de sódio em camadas, oferecendo orientação para projetar baterias mais duráveis e robustas.
Camadas atômicas empilhadas que precisam manter sua forma
Muitos cátodos promissores para íon-sódio são construídos a partir de pilhas de folhas atômicas planas. Íons de sódio ficam entre camadas de metal de transição-oxigênio em um arranjo ordenado “O3” quando totalmente sodiado, mas o carregamento e descarregamento repetidos empurram a estrutura para um padrão de empilhamento diferente, chamado “P3”. Essas mudanças em como as camadas se alinham — a sequência de empilhamento — podem ser reversíveis e inofensivas, ou podem desencadear colapso, fissuras e perda de capacidade. Os autores concentram-se em uma família de óxidos em camadas, Na(TM)O₂ com TM = Ti, Cr, Mn, Fe, Co ou Ni, e perguntam: quão fácil é para esses materiais reorganizarem seu empilhamento, e que papel as discordâncias desempenham quando isso acontece? 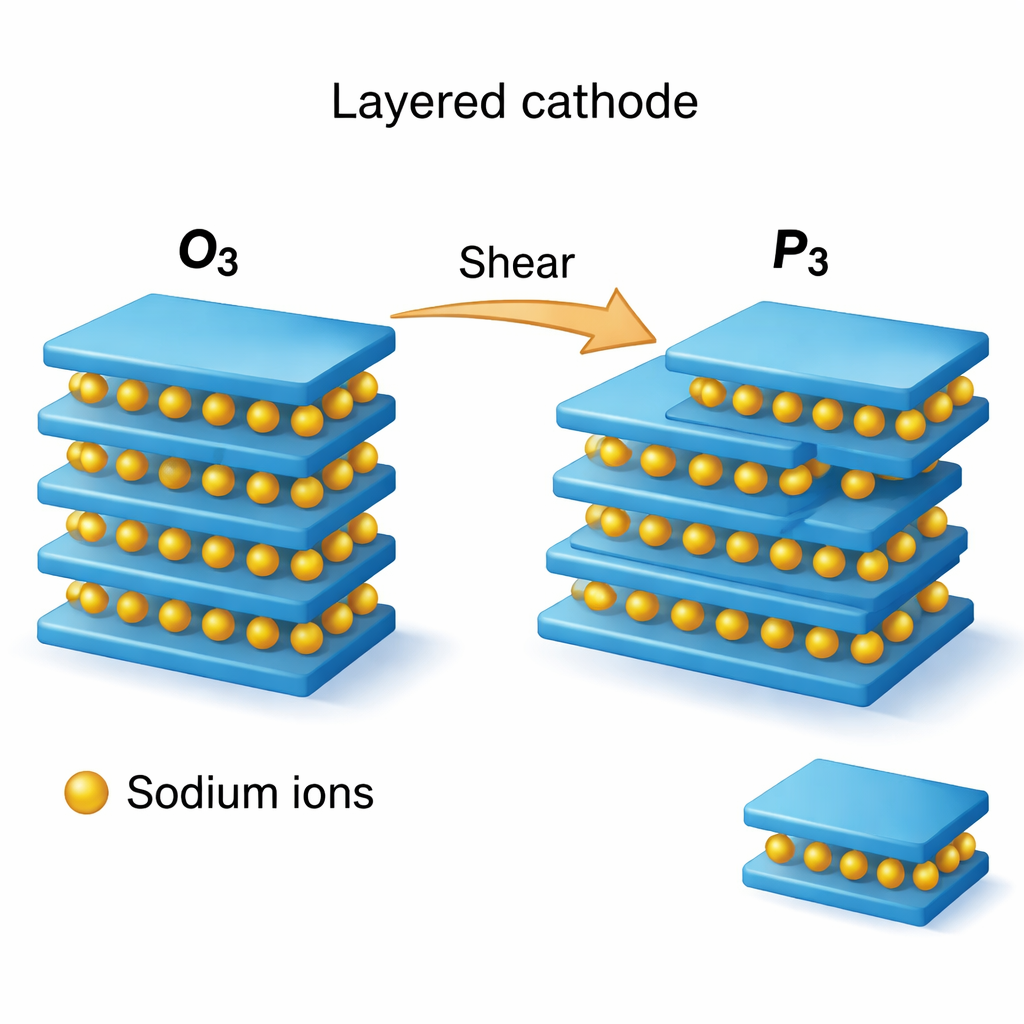
Mapeando como as camadas preferem deslizar
Para responder, os pesquisadores primeiro calculam as chamadas superfícies de energia de falha de empilhamento generalizada. Em termos simples, eles pegam duas metades do cristal, deslizam uma metade sobre a outra em diferentes direções e computam quanto cada deslocamento custa em energia. Caminhos de baixa energia nesse mapa revelam como as camadas preferem deslizar e se estados intermediários “fautados” — rearranjos locais do empilhamento — são prováveis de se formar. Em todos os compostos estudados, eles encontram que um estado fautado similar ao P3 é possível, mas é especialmente favorecido em materiais à base de cobalto e níquel, que mostram mínimos de energia profundos para essa configuração. Em contraste, um empilhamento mais drástico do tipo O1 não aparece como um estado estável nas condições que modelam, sugerindo que as mudanças mais suaves O3↔P3 são intrinsecamente mais acessíveis.
Como as discordâncias se parecem dentro desses cátodos
Cristais reais não cisalam como blocos perfeitamente rígidos; eles se deformam através do movimento de discordâncias. Usando um modelo semi-discreto de Peierls–Nabarro informado por seus dados quântico-mecânicos, os autores reconstruem a estrutura interna — ou “núcleo” — tanto de discordâncias de aresta quanto de parafuso no plano de escorregamento chave paralelo às camadas. Eles verificam que os núcleos das discordâncias são muito estreitos, com apenas alguns nanômetros de largura, confirmando que esses materiais são mecanicamente rígidos. Discordâncias de aresta tendem a se dividir em duas discordâncias “parciais” separadas por uma faixa fina que localmente apresenta empilhamento do tipo P3, especialmente em óxidos ricos em Co e Ni onde o estado P3 é energeticamente favorecido. Discordâncias de parafuso geralmente permanecem mais compactas, mas em algumas composições (novamente, notadamente Co e Ni) elas também podem se dividir e criar regiões estreitas semelhantes ao P3.
Com que facilidade defeitos se movem sob as tensões da bateria
Em seguida, o estudo estima a tensão de Peierls — a tensão mínima de cisalhamento necessária para iniciar o movimento de uma discordância através da rede. Essa quantidade atua como uma resistência microscópica ao escoamento para defeitos individuais. Para todos os materiais examinados, as tensões requeridas (alguns até algumas dezenas de megapascais) estão dentro da faixa de tensões esperadas quando íons de sódio são inseridos e extraídos durante o ciclo. Isso significa que o movimento de discordâncias não é apenas possível, mas provável sob condições operacionais realistas. As computações também mostram que algumas estruturas, particularmente variantes monoclínicas de óxidos de Mn e Ni, oferecem maior resistência a certos tipos de movimento de discordância porque seus caminhos de deslizamento de baixa energia preferidos são mais restritos. 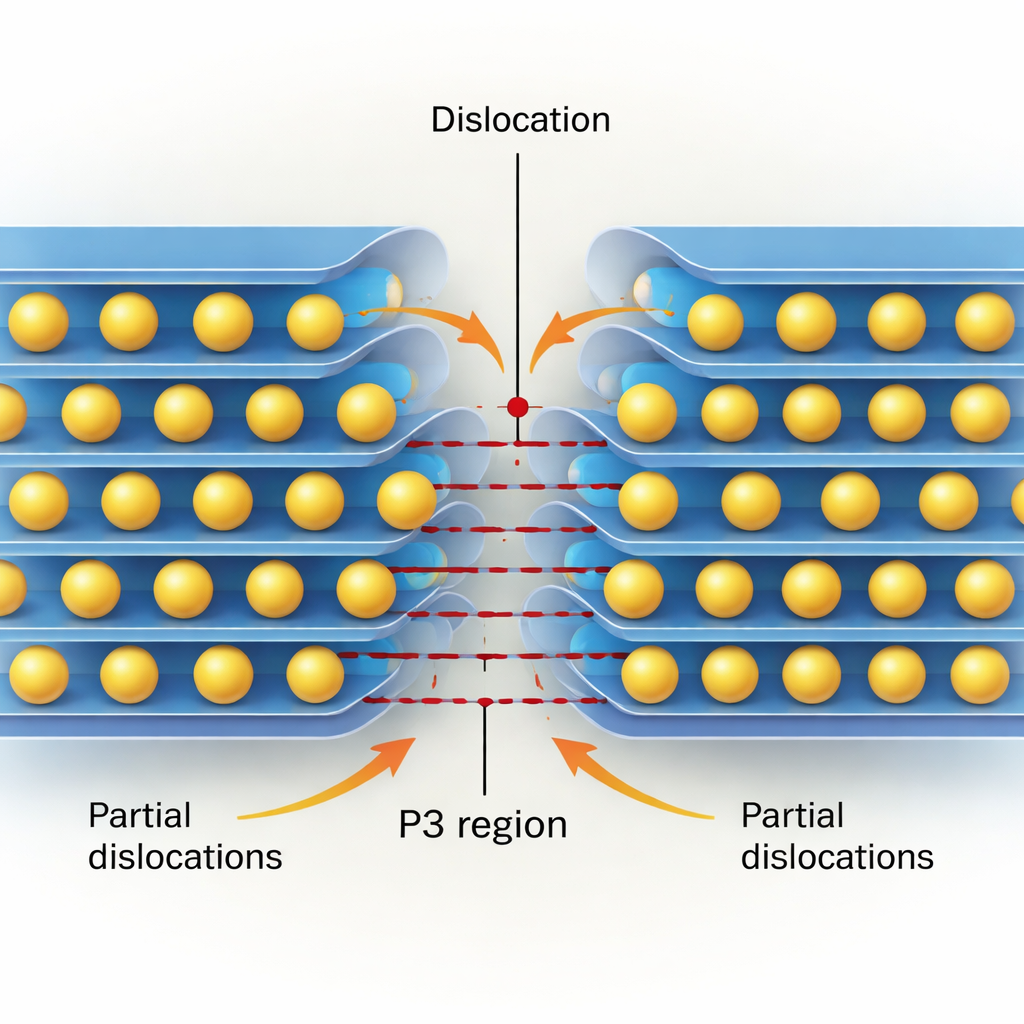
Discordâncias como motores de mudança de fase
Reunindo essas peças, os autores propõem um cenário em que as discordâncias impulsionam ativamente a transformação de fase O3→P3. Em um cátodo totalmente sodiado, discordâncias existentes ou recém-formadas podem se dividir em parciais, semeando pequenas regiões semelhantes ao P3 ao longo de sua linha. À medida que o sódio é removido, o panorama energético local muda de modo que a configuração P3 se torna cada vez mais estável. A faixa P3 entre as discordâncias parciais então se amplia, e os íons de sódio saltam para os novos sítios prismáticos, permitindo que a região P3 cresça e percorra a partícula. Ao longo de muitos ciclos, o acúmulo e o movimento desses defeitos também podem contribuir para microfissuras e fases irreversíveis, ligando processos em escala atômica diretamente à degradação da bateria.
Regras de projeto para baterias de sódio mais resistentes
Para um não especialista, a mensagem-chave é que a vida útil de baterias íon-sódio depende não apenas de quais elementos são escolhidos, mas também de como suas camadas atômicas preferem deslizar e de quão facilmente as discordâncias podem se mover. Ao mapear esses comportamentos a partir de primeira-princípios, o estudo fornece pistas de projeto: químicas que mantenham energias de falha de empilhamento rasas e controlem o movimento de discordâncias podem favorecer transições O3↔P3 suaves e reversíveis e resistir à formação de fissuras. Em termos práticos, isso significa que engenheiros podem ajustar composição e estrutura para gerenciar esses defeitos minúsculos, abrindo caminho para baterias íon-sódio mais baratas que as células de lítio de hoje, mas suficientemente duráveis para armazenamento de energia em larga escala.
Citação: Arcelus, O., Carrasco, J. First-principles computation of dislocation structures and stress-driven phase transformations in layered oxides for Na-ion batteries. npj Comput Mater 12, 96 (2026). https://doi.org/10.1038/s41524-026-01965-7
Palavras-chave: baterias íon-sódio, cátodos em camadas, discordâncias, transformações de fase, degradação de materiais