Clear Sky Science · pt
Uma mutação patogênica de Tau provoca disfunção autófia‑lisossomal que limita a degradação de Tau em um modelo de demência frontotemporal
Quando as equipes de limpeza do cérebro ficam para trás
Por que algumas pessoas desenvolvem problemas devastadores de memória e comportamento décadas antes da velhice? Este estudo aborda essa questão ao focalizar em uma única proteína cerebral, Tau, e nos minúsculos “centros de reciclagem” celulares que normalmente a mantêm sob controle. Observando neurônios humanos vivos com microscópios super‑potentes, os pesquisadores mostram como uma mutação de Tau causadora de doença entope o sistema de eliminação de resíduos da célula e como estimular esse sistema com uma pequena molécula pode ajudar a limpar a bagunça. As descobertas podem apontar para novas estratégias terapêuticas para certas formas de demência.
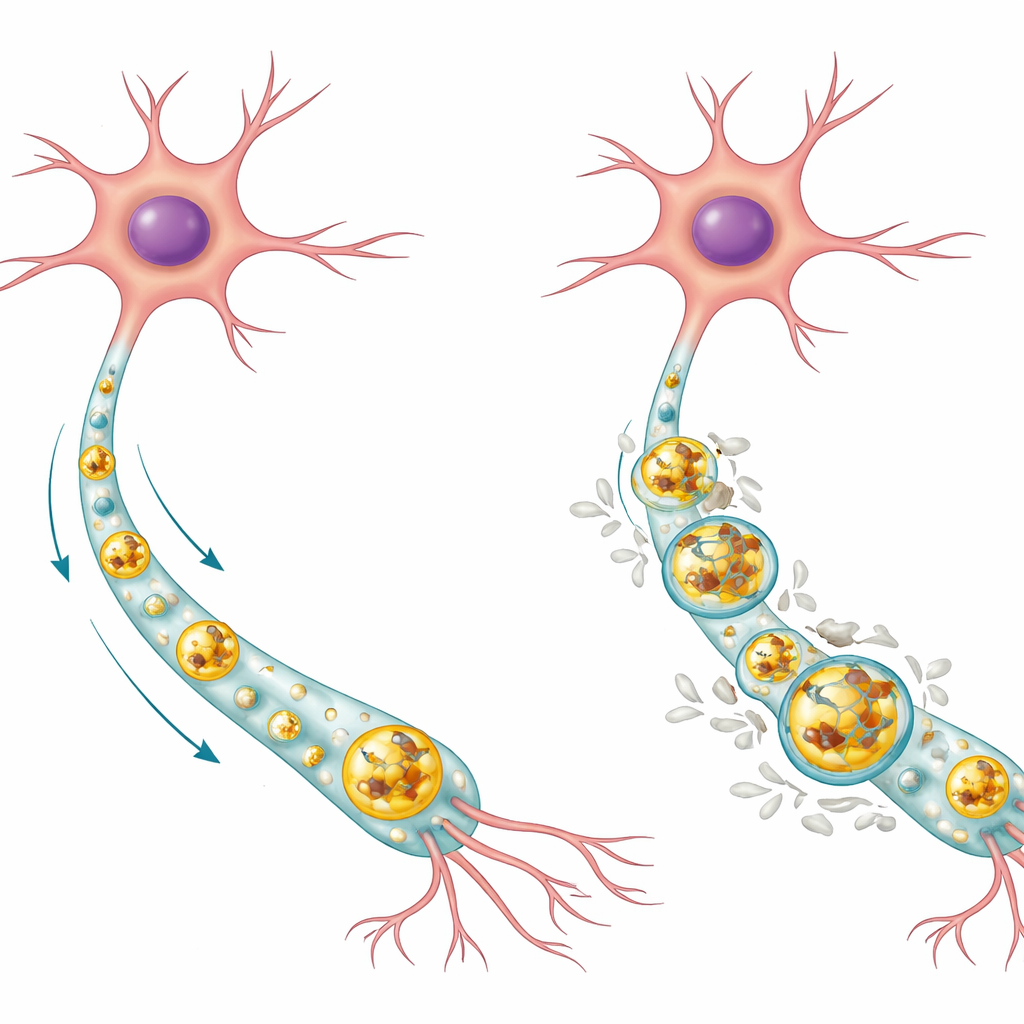
Como as células cerebrais normalmente tiram o lixo
Os neurônios são células de longa duração que não podem simplesmente se dividir para diluir material danificado, por isso dependem fortemente de sistemas internos de limpeza. Uma via chave é a via autofagia‑lisossomal. Nesse processo, proteínas indesejadas e partes desgastadas são envolvidas em sacos de membrana chamados autofagossomos, que então se fundem com compartimentos repletos de enzimas conhecidos como lisossomos, onde a carga é degradada e reciclada. Em neurônios humanos saudáveis, os autores descobriram que a proteína Tau normal tende a se acumular no centro ácido dos lisossomos, onde pode ser degradada, enquanto a forma fosforilada de Tau (uma modificação química ligada à doença) se localiza mais na membrana externa do lisossomo. A maioria dos lisossomos em células saudáveis estava totalmente livre de Tau, sugerindo que esse sistema normalmente mantém os níveis de Tau baixos e bem controlados.
O que dá errado em uma forma genética de demência
A equipe concentrou‑se em uma mutação no gene MAPT, chamada p.R406W, que causa uma forma hereditária de demência frontotemporal e pode imitar a perda de memória semelhante à do Alzheimer. Usando tecnologia de células‑tronco, reprogramaram células da pele de pacientes em células pluripotentes induzidas e, em seguida, em grande número de neurônios humanos que ou carregavam a mutação ou haviam sido corrigidos por edição gênica para o estado normal. Nos neurônios mutantes, Tau total e Tau fosforilada estavam marcadamente elevados, não porque as células produzissem mais Tau, mas porque o eliminavam de forma menos eficiente. Imagens de super‑resolução revelaram que quase todos os lisossomos nas células mutantes estavam lotados de Tau e, especialmente, com Tau fosforilada revestindo a membrana lisossomal. Esse acúmulo indicava que a principal via de eliminação de proteínas da célula estava entupida.
Centros de reciclagem entupidos e tráfego lento
Ao observar mais de perto a máquina de reciclagem, os pesquisadores perceberam que os lisossomos em neurônios mutantes eram mais numerosos, maiores e tendiam a ficar mais distantes do corpo celular. Imagens ao vivo com corantes fluorescentes mostraram que esses lisossomos se moviam mais lentamente e percorriam distâncias menores ao longo das fibras nervosas, embora as trilhas de microtúbulos subjacentes aparentassem normais. Os neurônios mutantes também continham mais autofagossomos, mais da proteína adaptadora de carga p62 e gotículas de lipídios extras — sinais de que material estava sendo marcado para descarte, mas não totalmente degradado. Usando um repórter sensível ao pH, descobriram que os autofagossomos nas células mutantes frequentemente falhavam em se fundir adequadamente com os lisossomos, levando a um acúmulo de vesículas de reciclagem “meio prontas” e a defeitos amplos na limpeza celular, não apenas para Tau, mas também para outras cargas.
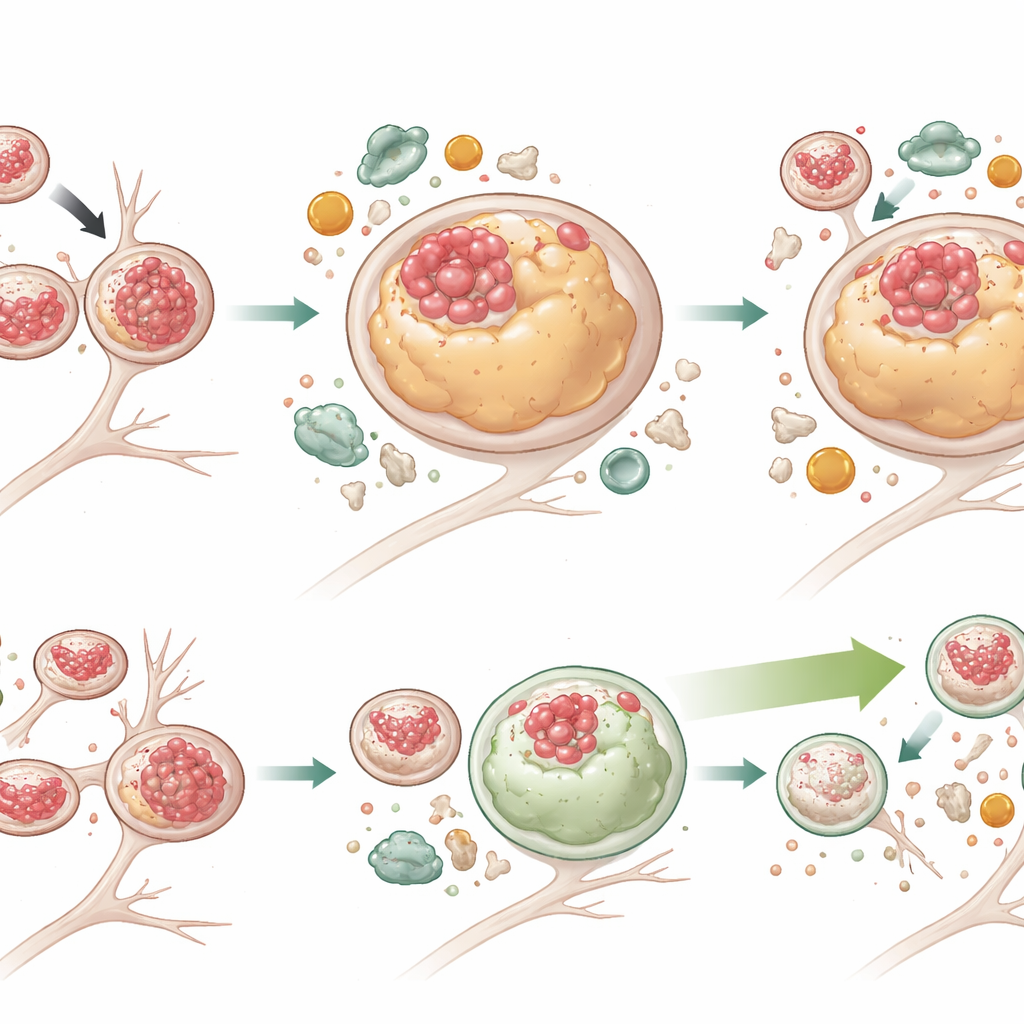
Impulsionando a limpeza celular sem consertar o engarrafamento
Para testar se aumentar a autofagia poderia superar esses problemas, a equipe tratou os neurônios com G2‑567, uma pequena molécula previamente demonstrada como estimulante do sistema autofagia‑lisossomal. Após duas semanas de tratamento, os neurônios mutantes exibiram níveis substancialmente mais baixos tanto de Tau total quanto de Tau fosforilada, e muitos mais lisossomos voltaram a ficar livres de Tau. Os lisossomos também encolheram em direção ao tamanho normal. Marcadores de autofagia ativa aumentaram, enquanto p62 — um indicador de degradação emperrada — diminuiu nas células mutantes, demonstrando uma degradação de carga mais eficaz. Curiosamente, G2‑567 não corrigiu todos os defeitos: os lisossomos em neurônios mutantes ainda tendiam a ficar mais distantes do corpo celular e a se mover lentamente, e uma proteína adaptadora (JIP3) ligada ao transporte lisossomal permaneceu elevada. Isso sugere que as funções de movimento e degradativas dos lisossomos podem ser parcialmente desacopladas, e que melhorar apenas a degradação pode ser suficiente para reduzir o acúmulo tóxico de Tau.
O que isso significa para futuros tratamentos da demência
Para um público não especializado, a conclusão principal é que, neste modelo genético de demência frontotemporal, o problema não é simplesmente que Tau se torna anormal; é que o sistema de reciclagem do neurônio não consegue dar conta. A mutação p.R406W em Tau interrompe diretamente várias etapas da via autofagia‑lisossomal, fazendo com que Tau — especialmente sua forma fosforilada — se acumule sobre e dentro dos lisossomos, junto com outro material não degradado. Ao empurrar farmacologicamente a máquina de limpeza celular para trabalhar mais, os pesquisadores conseguiram reduzir os níveis de Tau e normalizar o tamanho dos lisossomos, mesmo que os defeitos de transporte persistissem. Essas descobertas reforçam a ideia de que fármacos projetados para aumentar com segurança a autofagia e a função lisossomal poderiam ajudar a restaurar o equilíbrio proteico em demências relacionadas à tau e talvez em condições mais comuns, como a doença de Alzheimer.
Citação: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Palavras-chave: proteína tau, autofagia, disfunção lisossomal, demência frontotemporal, neurodegeneração