Clear Sky Science · pt
DiNovo possibilita sequenciamento de peptídeos de novo com alta cobertura e alta confiança por meio de proteases espelhadas e aprendizado profundo
Vendo proteínas com novo nível de detalhe
Proteínas são as pequenas máquinas que mantêm nossas células vivas, mas ler por completo seus blocos construtores ainda é surpreendentemente difícil. Este artigo apresenta o DiNovo, um novo sistema de software que ajuda cientistas a “ler” fragmentos proteicos de forma muito mais completa e confiável do que antes. Ao combinar um truque bioquímico engenhoso com inteligência artificial moderna, ele promete descobrir proteínas ocultas, marcadores de doenças e até alvos imunológicos que métodos tradicionais frequentemente deixam passar.
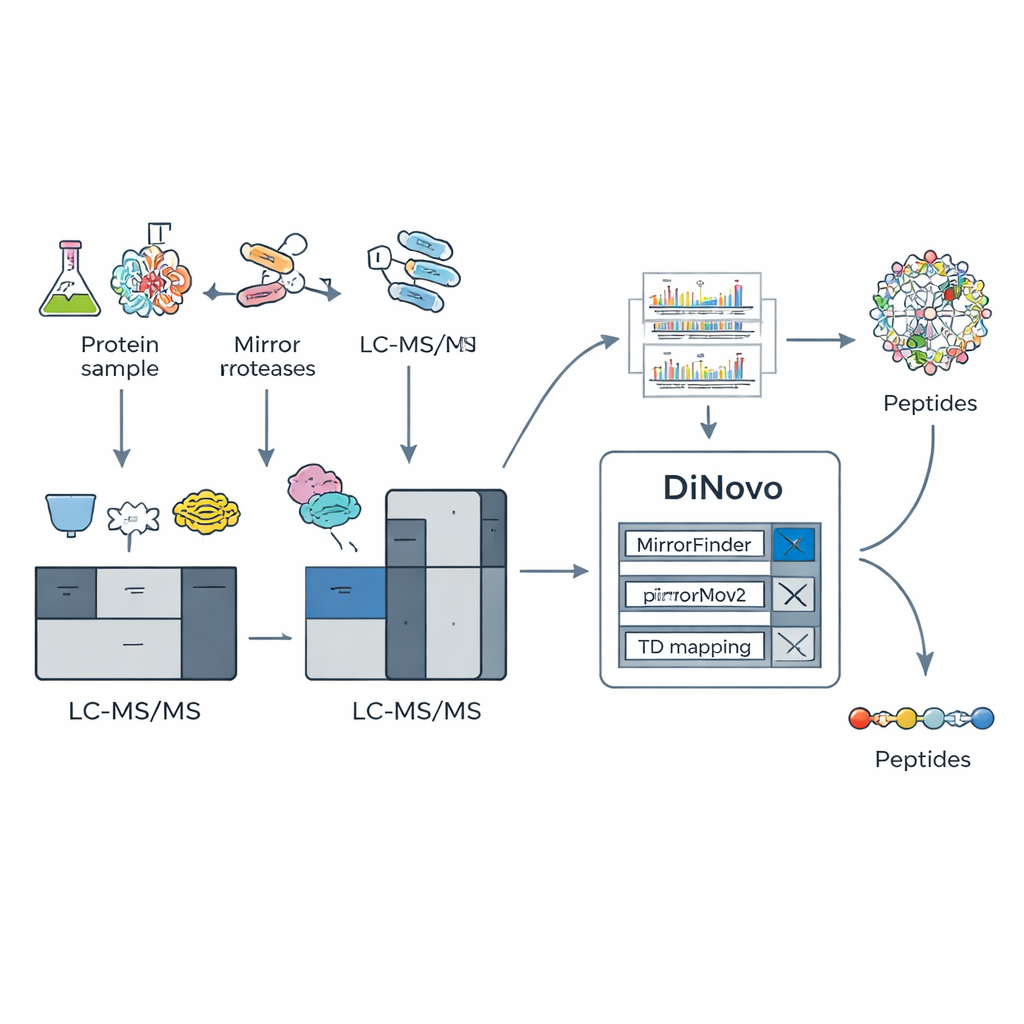
Por que ler fragmentos proteicos é tão difícil
A maioria das análises proteicas hoje depende de cortar proteínas em pedaços menores, chamados peptídeos, e depois pesar seus fragmentos em um espectrômetro de massas. A partir desses pesos, computadores tentam reconstruir a sequência original do peptídeo, como resolver um crucigrama a partir de pistas parciais. Métodos existentes costumam assumir que os peptídeos vêm de bancos de dados de proteínas conhecidos, o que funciona bem para proteínas familiares, mas tem dificuldade com proteínas novas ou inesperadas. O chamado sequenciamento de novo evita essa limitação ao tentar ler peptídeos diretamente a partir dos dados, mas frequentemente fica aquém porque alguns fragmentos estão ausentes e alguns peptídeos nunca são cortados de forma limpa em primeiro lugar.
Usando enzimas espelhadas para preencher as lacunas
A ideia central por trás do DiNovo é usar pares de “proteases espelhadas” – pares de enzimas de clivagem que cortam proteínas em lados opostos do mesmo tipo de aminoácido. Por exemplo, uma enzima corta logo antes de uma lisina, enquanto sua parceira corta logo depois dessa lisina. Isso produz dois peptídeos relacionados que compartilham o mesmo segmento interno, mas têm extremidades diferentes. Quando esses peptídeos “espelhados” são analisados, seus espectros de massa contêm padrões de fragmentos complementares: o que falta em um espectro muitas vezes aparece no outro. Os autores mostram que combinar tais pares espelhados pode levar a uma cobertura de fragmentos próxima do completo, com cerca de 98% dos cortes possíveis suportados por sinais experimentais reais, muito acima do observado ao usar apenas uma enzima.
Um pipeline de software inteligente feito para dados espelhados
Para explorar esse truque bioquímico, a equipe construiu o DiNovo como um fluxo de trabalho de software ponta a ponta. Primeiro, proteínas de bactérias e leveduras são digeridas com dois pares espelhados de enzimas, e os peptídeos resultantes são analisados por espectrometria de massas de alta resolução. O DiNovo então usa um módulo chamado MirrorFinder para reconhecer automaticamente quais pares de espectros vêm de peptídeos espelhados, fazendo isso diretamente a partir dos padrões de sinal em vez de quaisquer suposições de sequência prévias. Em seguida, seu motor principal de novo, MirrorNovo, utiliza aprendizado profundo para interpretar esses espectros pareados, enquanto um motor de reserva baseado em grafos, pNovoM2, oferece uma opção mais rápida que roda apenas em CPU. Juntos, essas ferramentas traduzem picos em sequências de aminoácidos e também examinam os espectros individuais que não formaram pares óbvios, extraindo o máximo de informação possível.
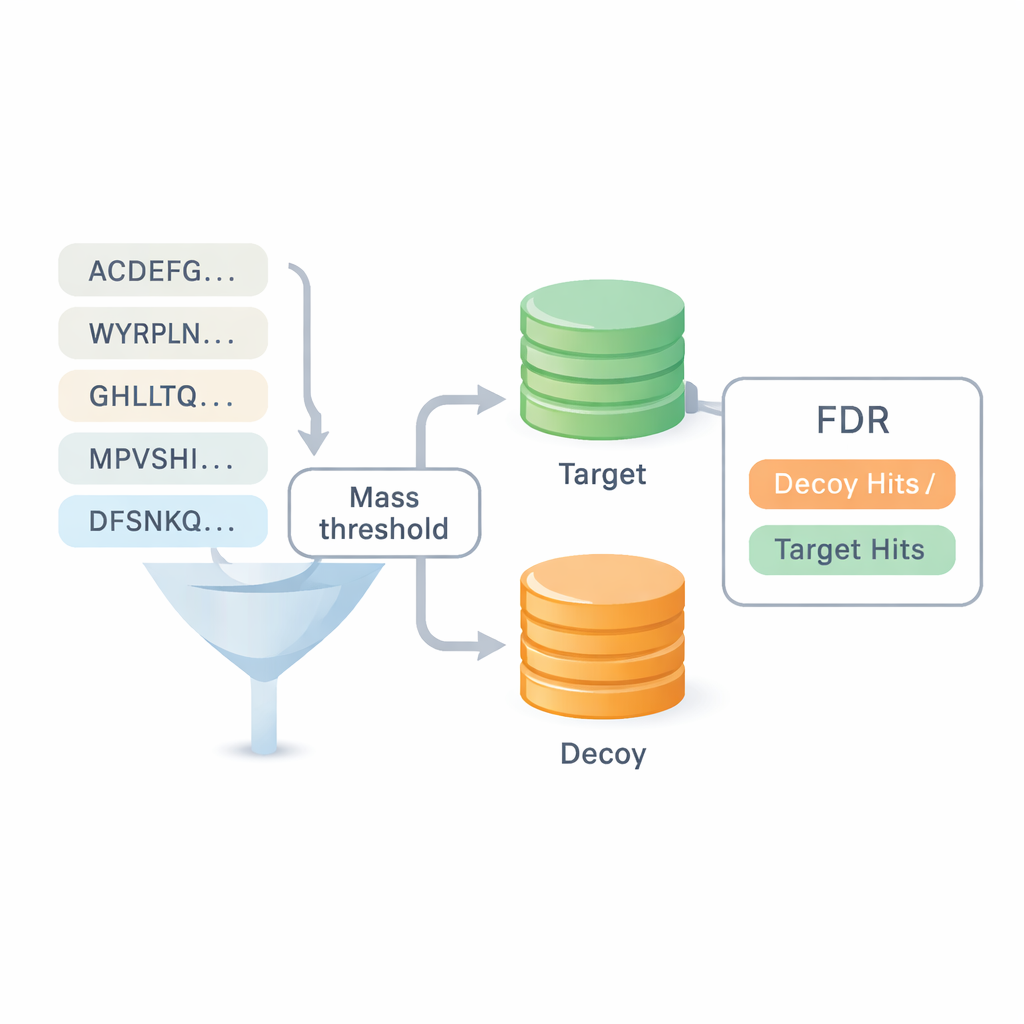
Medindo confiança sem depender de bancos de dados antigos
Uma das maiores questões no sequenciamento de novo é o quanto confiar nos resultados. A maioria dos benchmarks existentes recicla respostas de buscas em bancos de dados, o que borroneia a linha entre as duas abordagens e pode esconder erros. O DiNovo introduz um método de verificação de qualidade diferente chamado mapeamento alvo-engodo (target-decoy mapping). Aqui, os peptídeos recém-lidos são mapeados para uma coleção combinada de sequências de proteínas reais (alvo) e artificiais embaralhadas (engodo). Ao comparar com que frequência peptídeos caem no conjunto real versus no conjunto embaralhado, o software pode estimar uma taxa de erro, ou taxa de descoberta falsa, sem se apoiar em identificações anteriores. Isso torna possível comparar o DiNovo diretamente com programas padrão de busca em banco de dados sob os mesmos controles de erro.
O que o DiNovo entrega na prática
Em testes com amostras bacterianas, de levedura e anticorpos, o DiNovo consistentemente leu muito mais peptídeos e aminoácidos do que ferramentas de novo conhecidas que usam apenas uma enzima. Usando dois pares espelhados, ele produziu de 2 a 3 vezes mais aminoácidos de alta confiança do que uma configuração clássica apenas com tripsina e identificou mais proteínas em níveis de erro semelhantes. Quando comparado diretamente a três principais motores de busca em banco de dados, o DiNovo encontrou números semelhantes de aminoácidos e proteínas, e a maioria de suas sequências concordou com as dos motores de busca nos mesmos espectros. Os autores argumentam que esse nível de cobertura e concordância significa que o sequenciamento de novo, há muito tratado como método secundário, pode agora ficar ao lado da busca em banco de dados como uma opção séria e, em alguns casos, superior.
Panorama geral: rumo a uma leitura proteica completa e imparcial
Para um não especialista, a conclusão é que o DiNovo torna muito mais fácil ler pedaços de proteína com precisão sem ficar limitado ao que já existe em bancos de referência. Ao dobrar ou triplicar a quantidade de informação de sequência bem suportada e fornecer suas próprias verificações de erro integradas, essa abordagem abre portas para descobrir proteínas desconhecidas, rastrear variações sutis e explorar misturas complexas onde muitos componentes ainda são desconhecidos. Em suma, ao emparelhar enzimas espelhadas com aprendizado profundo e estatística cuidadosa, o DiNovo ajuda a transformar traços espectrais ruidosos em uma imagem mais clara e confiável das proteínas que sustentam a saúde e a doença.
Citação: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Palavras-chave: proteômica, sequenciamento de peptídeo de novo, espectrometria de massas, aprendizado profundo, proteases espelhadas