Clear Sky Science · pt
Amostragem eficiente de caminhos de transição em grande escala e conformações intermediárias em complexos proteicos sub-mesoscópicos
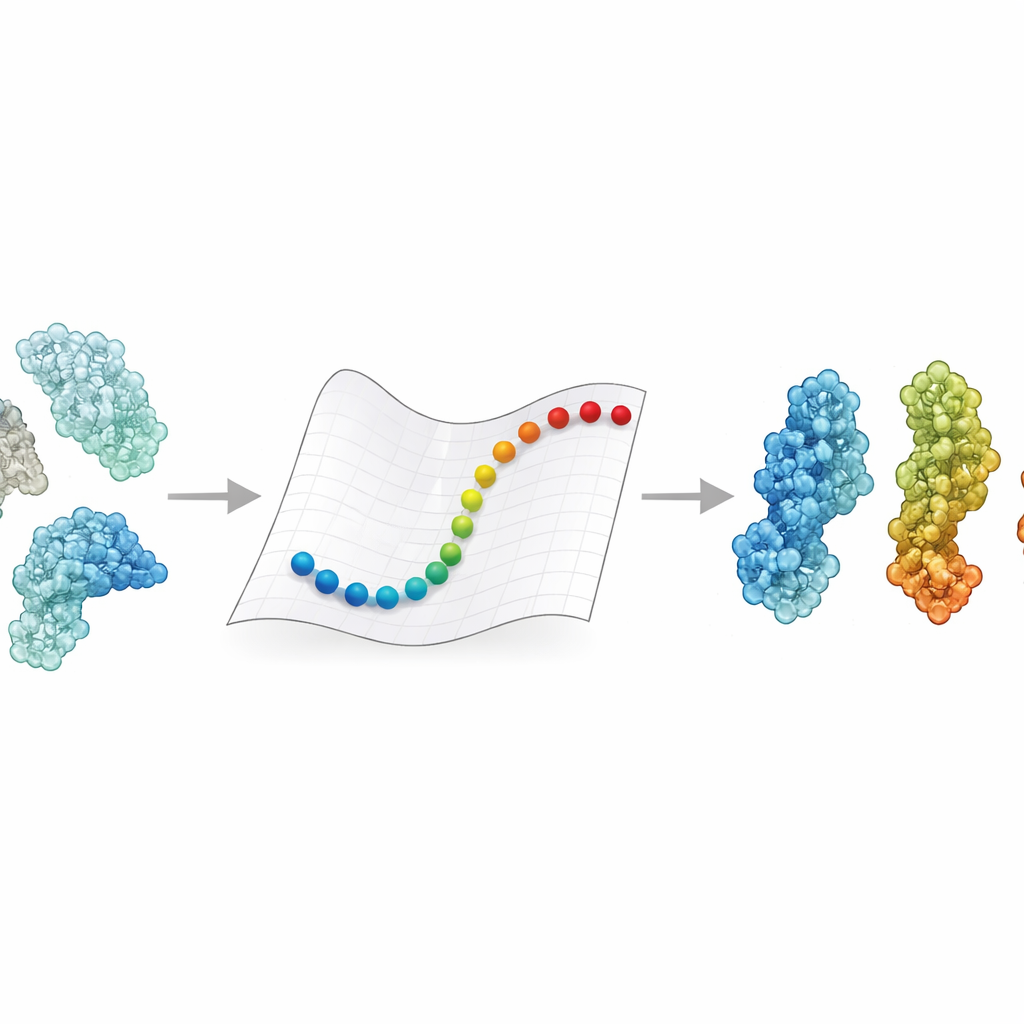
Observando proteínas em movimento
Muitas das moléculas que nos mantêm vivos comportam-se menos como peças rígidas de Lego e mais como pequenas máquinas que mudam constantemente de forma. Esses movimentos impulsionam processos como produção de energia, reparo de DNA e a entrada de vírus nas células. Experimentos como a crio‑microscopia eletrônica conseguem hoje congelar algumas dessas conformações, mas não os passos fugazes entre elas. Este artigo apresenta o eBDIMS2, um novo método computacional que pode “preencher os quadros faltantes” do movimento protéico, mesmo para máquinas moleculares enormes que antes eram grandes e complexas demais para serem simuladas em um computador comum.
Por que as mudanças de forma das proteínas importam
Proteínas raramente permanecem travadas em uma única posição. Elas se abrem e fecham, torcem e dobram em resposta a sinais como variações de voltagem, pH ou ao acoplamento de uma molécula parceira. Essas alterações podem determinar se uma enzima está ativa ou inativa, ou se um receptor prende um vírus ou o deixa escapar. Experimentos nos dão instantâneos detalhados de algumas conformações-chave, e simulações de dinâmica molecular podem em princípio conectá‑las acompanhando cada átomo ao longo do tempo. Mas seguir esse movimento para os enormes complexos hoje observados por crio‑ME — frequentemente com centenas de milhares a milhões de Daltons — geralmente exige supercomputadores e semanas de processamento. Como resultado, para muitos desses gigantes de interesse médico, ainda não sabemos como um estado se transforma em outro.
Uma rota mais rápida pelos paisagens protéicas
O eBDIMS2 faz um atalho ao simplificar como as proteínas são representadas e como seu movimento é calculado. Em vez de acompanhar cada átomo, trata cada aminoácido como um único ponto conectado por molas em uma rede elástica. Essas molas capturam como diferentes partes da proteína tendem a se mover em conjunto. O método então usa dinâmica Browniana — regras matemáticas que imitam o tremeluzir em um líquido — para empurrar a estrutura de um estado experimentalmente conhecido para outro. Crucialmente, o eBDIMS2 presta atenção apenas às interações que realmente importam, usando cortes de distância e computação paralela para reduzir o custo. Isso melhora a escalabilidade do programa de aproximadamente quadrática para quase linear em relação ao tamanho da proteína. Na prática, isso significa que transições para conjuntos enormes, próximos a dois milhões de Daltons, podem ser exploradas em horas em um computador de mesa, em vez de serem praticamente inacessíveis.
Comparando os caminhos com proteínas reais
Para verificar se esses caminhos rápidos fazem sentido biológico, os autores reuniram conjuntos de 47 proteínas grandes e 15 complexos adicionais, totalizando centenas de estruturas em sua maioria resolvidas por crio‑microscopia eletrônica. Eles usaram análise de componentes principais, uma ferramenta estatística que destaca as maneiras dominantes pelas quais cada proteína pode se mover, para organizar essas estruturas em paisagens conformacionais como aberto, fechado, ativo ou inativo. Em seguida, pediram ao eBDIMS2 que conectasse pares de estados finais nessa paisagem. Os caminhos resultantes foram projetados de volta nos mesmos mapas de baixa dimensionalidade, revelando se traçavam rotas suaves que passavam perto de intermediários observados experimentalmente. Em mais de 30% dos sistemas, as rotas simuladas passaram perto — a poucos ângströms — de estruturas intermediárias que não haviam sido fornecidas como entrada. Para casos exigentes, como a enzima reparadora de DNA DNA‑PKcs ou a proteína spike do coronavírus, os caminhos coarse-grained também se sobrepuseram bem a simulações muito mais caras ao nível atômico, incluindo dinâmica molecular dirigida e execuções avançadas de amostragem aprimorada.
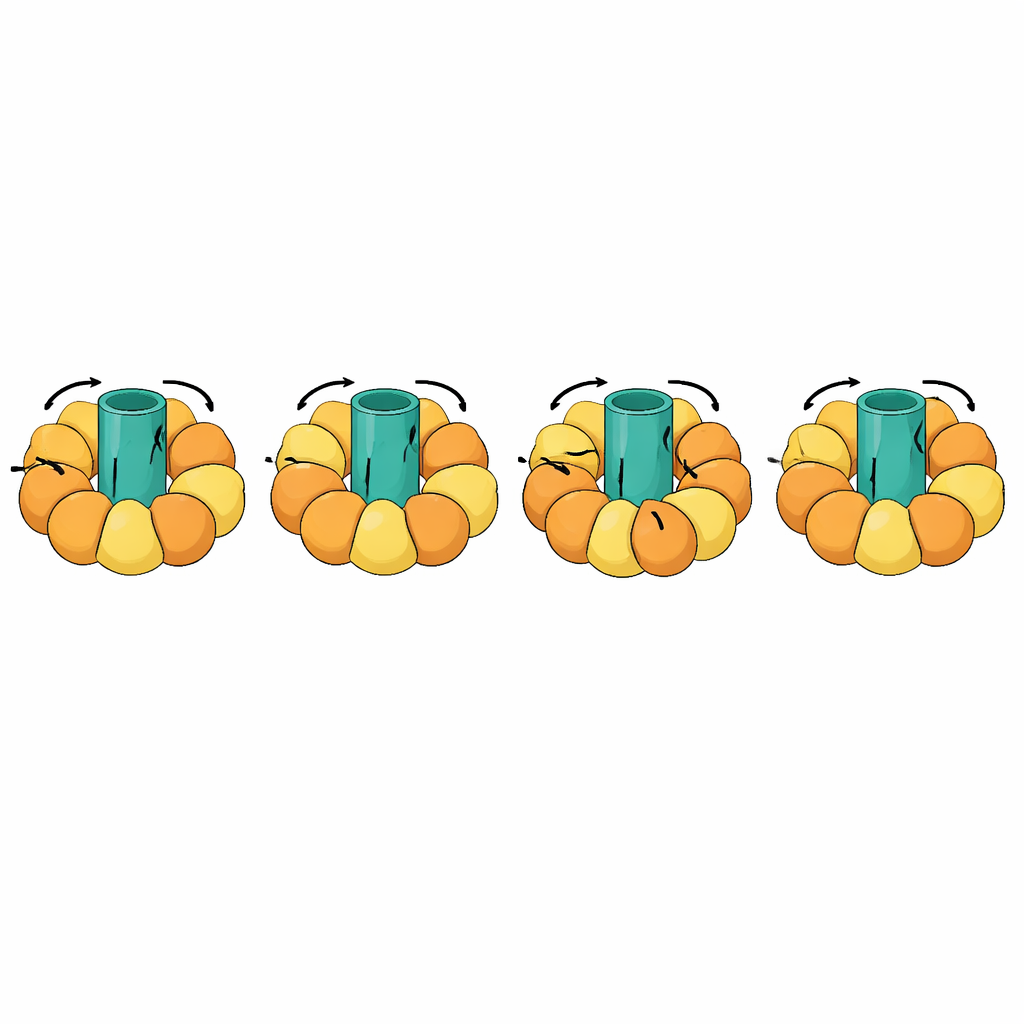
Acompanhando máquinas moleculares gigantes
Um dos testes mais impressionantes envolveu máquinas rotativas como as ATP sintases, que produzem a moeda energética da célula ao acoplar um rotor giratório na membrana a movimentos de abertura e fechamento em subunidades circundantes. Essas transições são excepcionalmente complexas: partes da molécula devem permanecer rígidas e girar como uma unidade, enquanto outras se flexionam em um ciclo coreografado. O eBDIMS2 introduz tratamento especial para tais peças quase‑rígidas e para modelos experimentais incompletos com segmentos ausentes, ambos comuns em crio‑ME. Com esses recursos, ele pode simular ciclos rotacionais completos de ATP sintase e de outros complexos massivos como chaperonas moleculares, receptores e agregados virais. Em todos os casos, as estruturas intermediárias geradas evitam as fortes distorções produzidas por alguns métodos concorrentes e podem ser convertidas em modelos atomísticos adequados para cálculos de design de fármacos ou para simulações mais longas e detalhadas.
O que isso significa para a biologia e a medicina
O estudo mostra que o eBDIMS2 pode esboçar de forma confiável as rotas principais entre conformações proteicas conhecidas para sistemas que estavam fora do alcance das simulações tradicionais. Ele não substitui filmes detalhados ao nível atômico nem fornece energias e cronogramas precisos, mas oferece uma maneira rápida e fisicamente fundamentada de mapear como grandes máquinas moleculares podem se mover, usando apenas um par de estruturas experimentais como entrada. À medida que bancos de dados estruturais se enchem com múltiplos estados de grandes complexos proteicos ligados ao câncer, infecções e outras doenças, essa abordagem dá aos pesquisadores uma ferramenta acessível para conectar pontos, sugerir estados intermediários plausíveis e orientar onde procurar a seguir com métodos de maior resolução ou com design de fármacos direcionado.
Citação: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Palavras-chave: dinâmica de proteínas, simulações moleculares, crio‑ME, caminhos conformacionais, modelagem grosseira (coarse-grained)