Clear Sky Science · pt
Ativação de IRF3 em cardiomiócitos prejudica a função oxidativa mitocondrial por inibição de PGC-1α e impulsiona a insuficiência cardíaca
Por que corações estressados e células cansadas importam
A insuficiência cardíaca costuma ser descrita como o coração “se desgastando”, mas por trás disso também há uma história de inflamação crônica e usina de energia exaurida dentro das células do músculo cardíaco. Este estudo faz uma pergunta aparentemente simples, com grandes implicações: existe um único interruptor molecular dentro das células cardíacas que conecta inflamação prejudicial e falha na produção de energia — e, em caso afirmativo, mudar esse interruptor pode alterar o curso da insuficiência cardíaca? Seguindo esse fio, os autores identificam um ator-chave e mostram que um reforço moderado do próprio programa energético do coração pode resgatar parcialmente corações em falha em camundongos.
Um interruptor molecular em corações humanos doentes
Os pesquisadores concentraram-se em uma proteína chamada IRF3, mais conhecida por ajudar as células a responder a infecções virais. Eles examinaram tecido de pessoas com cardiomiopatia isquêmica, uma forma comum de insuficiência cardíaca causada por fluxo sanguíneo reduzido após infartos. Nesses corações em falha, o IRF3 não estava apenas presente — ele estava quimicamente ativado em locais específicos, sinal de que estava dirigindo programas gênicos. Ao mesmo tempo, a maquinaria que permite às mitocôndrias transformar combustível em energia por fosforilação oxidativa estava visivelmente enfraquecida. Um padrão semelhante apareceu em modelos de camundongo de infarto: quando uma artéria coronária foi ocluída, o IRF3 nos cardiomiócitos foi fortemente ativado, e genes controlados por IRF3 foram induzidos. Mesmo fragmentos de DNA mitocondrial — liberados de mitocôndrias danificadas e atuando como sinais internos de “perigo” — foram suficientes para ativar o IRF3 em células cardíacas isoladas.
Desligar o IRF3 protege o coração
Para testar se a atividade do IRF3 nos cardiomiócitos realmente piora a doença, a equipe criou camundongos nos quais o IRF3 podia ser removido apenas dos cardiomiócitos, deixando intactas outras células imunes e de suporte. Após induzir um infarto, esses camundongos apresentaram melhor função de bombeamento e menos cicatrização do que camundongos normais, apesar de terem a mesma lesão inicial. Em células cardíacas cultivadas, silenciar o IRF3 reduziu a expressão de genes inflamatórios sem perturbar outras proteínas relacionadas. Juntos, esses resultados indicam que o IRF3 dentro da própria célula cardíaca não é apenas um espectador: ele amplifica a inflamação e o dano estrutural após isquemia e contribui para a transição para insuficiência cardíaca.
Quando o IRF3 fica “ligado”, o sistema de combustível colapsa
Os autores inverteram então o experimento: criaram camundongos nos quais o IRF3 nos cardiomiócitos podia ser forçado a um estado permanentemente ativo usando um truque genético “fosfomimético”. Mesmo sem um gatilho externo, esses animais desenvolveram rapidamente disfunção cardíaca severa, altos níveis de mensageiros inflamatórios no sangue e sinais de lesão celular. Uma análise aprofundada do tecido cardíaco mostrou que, quando o IRF3 está cronicamente ativo, ele suprime um coordenador mestre de energia chamado PGC-1α. Essa molécula normalmente promove mitocôndrias saudáveis, queima eficiente de lipídios e equilíbrio energético celular. Com PGC-1α reduzido, várias proteínas mitocondriais diminuíram, a cadeia de transporte de elétrons falhou e as escolhas de combustível do coração mudaram: carnitina e compostos relacionados para oxidação de gordura caíram, o uso de corpos cetônicos foi prejudicado e o manejo da glicose ficou distorcido. Até a razão NAD⁺/NADH — um indicador chave do balanço redox celular — inclinou-se na direção errada.
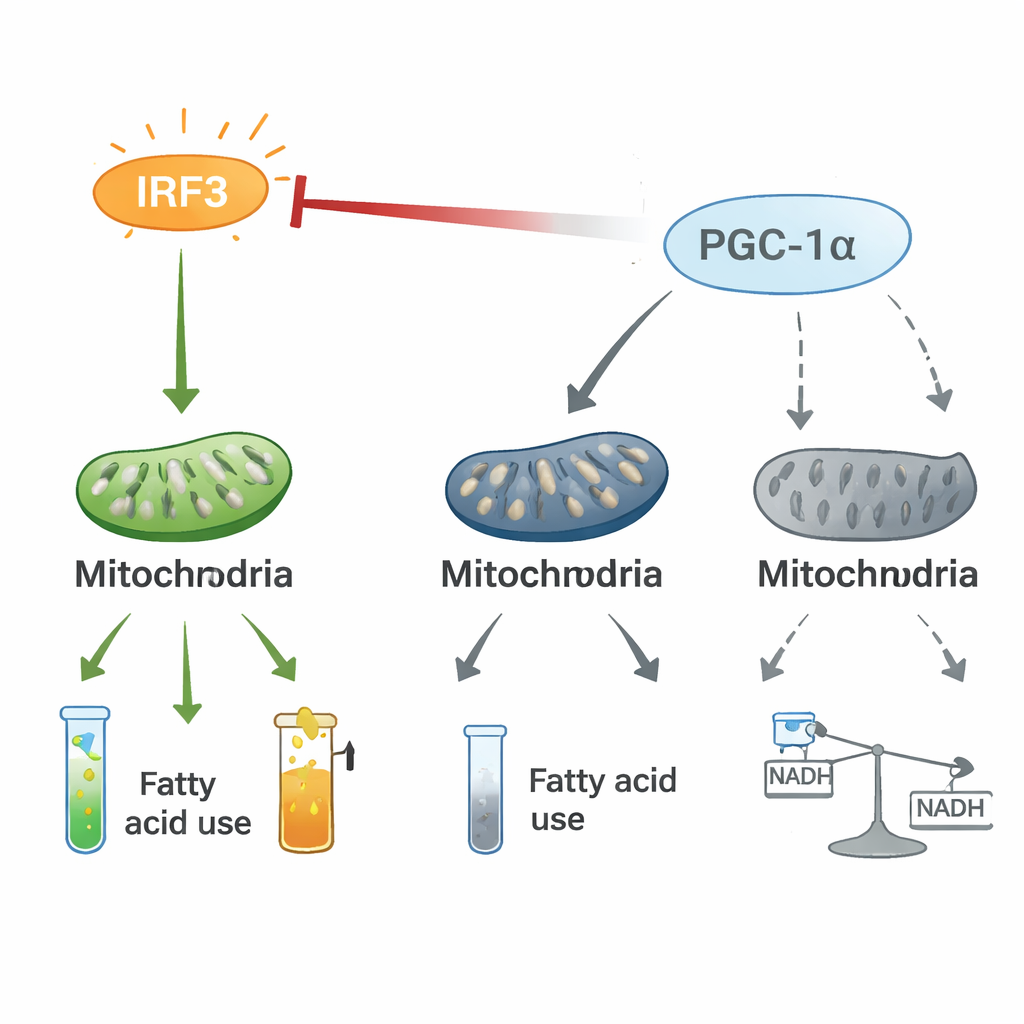
Uma disputa entre inflamação e controle energético
Experimentos mecanísticos revelaram que IRF3 e PGC-1α formam um eixo regulatório bidirecional. Em cardiomiócitos, o IRF3 ativado associa-se fisicamente ao PGC-1α e atenua sua capacidade de ativar genes de oxidação de gordura. Reduzir o IRF3 eleva os níveis e a atividade de PGC-1α, enquanto aumentar o PGC-1α suprime genes inflamatórios dirigidos por IRF3 e restaura marcadores mitocondriais, mesmo sob condições de estresse como hipóxia ou toxinas bacterianas. Rastreamento com isótopos estáveis mostrou que a ativação do IRF3 redireciona carbono do fluxo energético normal através do ciclo do ácido cítrico para uma via alternativa, a via das pentoses fosfato, e interrompe o fluxo regular de metabólitos. Essa disputa entre um interruptor pró-inflamatório (IRF3) e um copiloto energético (PGC-1α) parece remodelar o metabolismo cardíaco de formas que favorecem inflamação e perda de energia.
Recarregando suavemente as baterias do coração
Por fim, a equipe perguntou se empurrar o PGC-1α de volta para cima poderia contrariar o dano causado pelo IRF3. Eles usaram um vetor de terapia gênica com alvo cardíaco para aumentar moderadamente — mas não excessivamente — o PGC-1α nos mesmos camundongos com IRF3 hiperativo. Esse impulso modesto melhorou a função de bombeamento, aumentou proteínas mitocondriais, realçou genes de oxidação de gordura e do metabolismo do NAD, e reduziu a atividade de genes inflamatórios e fibróticos. Em experimentos celulares, coexpressar PGC-1α com IRF3 ativo restaurou um equilíbrio NAD⁺/NADH mais saudável e orientou novamente o uso de combustível para as gorduras. Para o leitor leigo, isso significa que recarregar com cuidado o “sistema de gerenciamento de bateria” do coração pode compensar parcialmente os efeitos nocivos de um interruptor inflamatório crônico preso na posição “ligado”.
O que isso significa para os cuidados futuros com a insuficiência cardíaca
Este trabalho posiciona o IRF3 como um elo central entre inflamação e falha energética dentro dos cardiomiócitos. Em vez de tratar inflamação e metabolismo como problemas separados na insuficiência cardíaca, o estudo sugere que eles estão interligados por um eixo IRF3–PGC-1α. Embora essas descobertas sejam em camundongos e células, elas levantam a possibilidade de que terapias futuras possam reduzir a atividade do IRF3 ou reforçar o PGC-1α e a função mitocondrial para desacelerar ou prevenir a insuficiência cardíaca após um infarto. Em termos simples, acalmar um sistema de alarme celular hiperativo e apoiar as usinas de energia do coração pode ser uma estratégia combinada poderosa para manter corações enfraquecidos batendo com mais força por mais tempo.
Citação: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Palavras-chave: insuficiência cardíaca, inflamação, mitocôndrias, cardiomiócitos, PGC-1α