Clear Sky Science · pt
XL-MSDigger: uma solução versátil baseada em aprendizado profundo para espectrometria de massa com entrecruzamento
Vendo como as proteínas se mantêm juntas
Cada processo em nossos corpos depende não só das proteínas se dobrarem nas formas corretas, mas também de encontrarem os parceiros certos. Ainda assim, observar essas relações moleculares em ação é notoriamente difícil. Este estudo apresenta o XL-MSDigger, uma plataforma de software que usa inteligência artificial moderna para extrair sinais muito mais claros de uma técnica experimental ruidosa chamada espectrometria de massa com entrecruzamento, ajudando cientistas a mapear como as proteínas estão organizadas e com quem interagem dentro das células.
Desembaralhando um mundo molecular lotado
Para entender como as proteínas são montadas e como elas se conectam, pesquisadores frequentemente usam espectrometria de massa com entrecruzamento. Nessa abordagem, pequenas “pontes” químicas conectam partes próximas das proteínas. Os pedaços ligados são então quebrados em fragmentos e pesados em um espectrômetro de massa. Em princípio, o padrão de fragmentos revela quais trechos de proteína estavam próximos no espaço, como descobrir quais páginas de um livro foram presas juntas. Na prática, entretanto, os dados resultantes são extremamente complexos. As ferramentas computacionais existentes costumam considerar apenas a informação básica de massa e têm dificuldade com o enorme número de combinações possíveis, levando a conexões perdidas e correspondências espúrias.
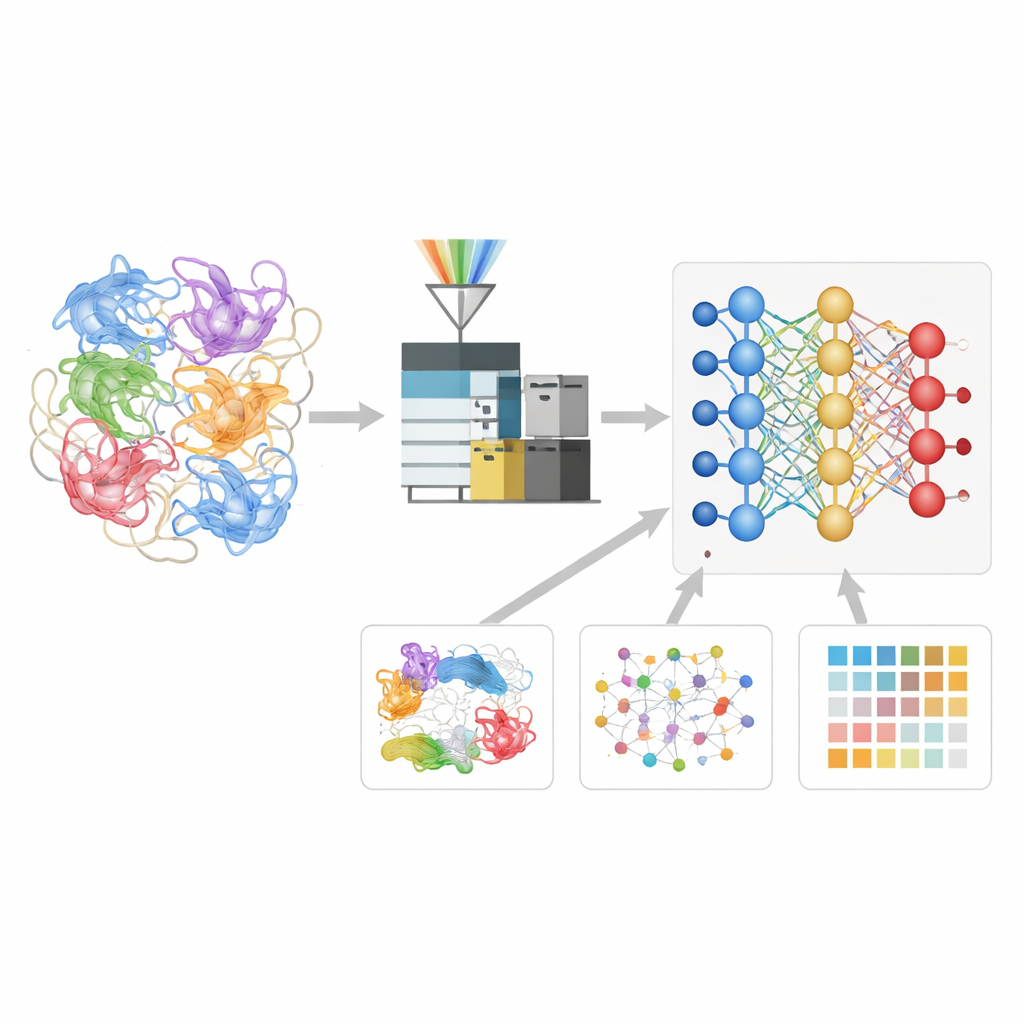
Ensinando uma rede neural a linguagem dos fragmentos proteicos
Os autores construíram um modelo de aprendizado profundo chamado Deep4D-XL para interpretar melhor esses experimentos de entrecruzamento. Primeiro, eles criaram um grande conjunto de referência ao entrecruzar proteínas de células humanas, quebrá-las em peptídeos e registrar não apenas suas massas, mas também quanto tempo levaram para percorrer o instrumento e como se comportaram em uma câmara de mobilidade iônica. Cada par entrecruzado foi codificado para o modelo, que usa um desenho em “siamês” duplo para ler ambos os parceiros peptídicos e um passo de atenção cruzada para combinar suas informações. A partir disso, a rede aprende a prever três propriedades-chave de qualquer novo peptídeo entrecruzado: quando deve aparecer no experimento, como deve se mover e como seu padrão de fragmentação deve ser.
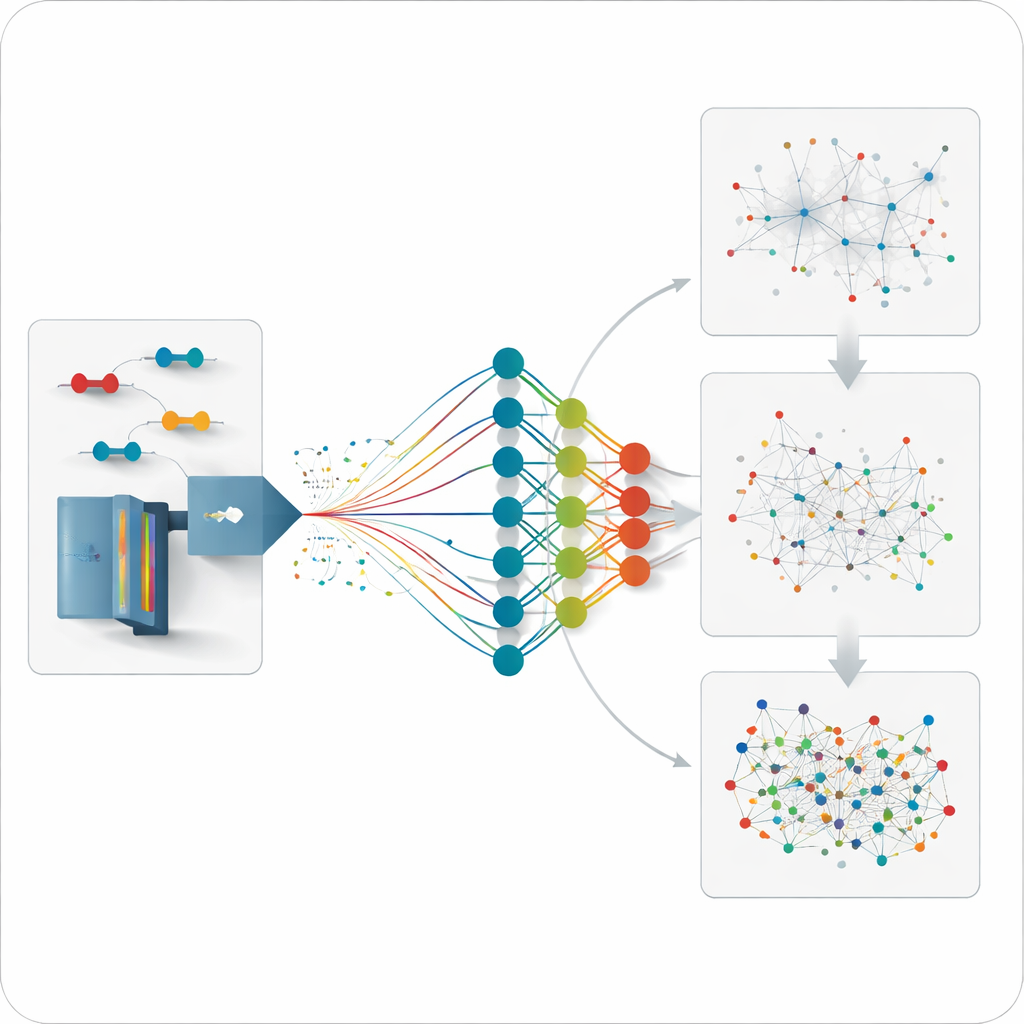
Convertendo previsões em sinais mais limpos
O XL-MSDigger envolve esse motor de predição em fluxos de trabalho analíticos para dois estilos principais de coleta de dados. No estilo tradicional, dirigido, o instrumento registra seletivamente fragmentos de íons que escolhe em tempo real. O XL-MSDigger pega as correspondências iniciais de softwares de busca estabelecidos e as reavalia usando o comportamento previsto pelo modelo para cada candidato. Uma segunda rede neural compara predição e experimento ao longo de várias dimensões e atribui pontuações aprimoradas. Essa reavaliação quase dobra o número de ligações entre diferentes proteínas detectadas com confiança em amostras de levedura e humanas, mantendo baixas as taxas de erro e revelando muitas mais interações proteína–proteína do que antes.
Fazendo sentido de enxurradas de dados sem viés
Uma forma mais nova de operar esses instrumentos, chamada aquisição independente de dados, registra fragmentos de quase tudo em uma amostra, melhorando a cobertura mas gerando dados avassaladores. Até agora, não havia uma boa maneira de estimar quantos dos entrecruzamentos resultantes eram realmente reais. O XL-MSDigger usa o Deep4D-XL para construir uma biblioteca “enganadora” (decoy) cuidadosamente pareada de entrecruzamentos falsos e então analisa entradas reais e decoys em conjunto. Ao observar com que frequência os decoys passam, o software pode estimar a taxa de descobertas falsas e treinar outra rede neural para separar correspondências verdadeiras das falsas. Essa reclassificação aumenta aproximadamente cinco vezes o número de sinais entrecruzados confiáveis e produz separação clara entre padrões reais e decoys.
Prevendo o que ainda não foi medido
Porque o modelo pode prever como qualquer peptídeo entrecruzado plausível deve se comportar, a equipe pode ir um passo além e analisar dados para ligações que nunca foram medidas diretamente antes. Eles geram bibliotecas previstas de tamanho moderado focadas em proteínas selecionadas ou redes de interação e então pesquisam os dados não direcionados contra essas bibliotecas. Essa estratégia descobre ligações adicionais dentro de proteínas individuais e entre parceiros de importantes proteínas chaperonas, com distâncias que concordam bem com estruturas tridimensionais conhecidas. Também recupera interações perdidas pelas bibliotecas experimentais tradicionais, mais limitadas, especialmente para conexões de baixa abundância.
Abrindo uma janela mais clara sobre parcerias proteicas
Para não especialistas, a mensagem-chave é que o XL-MSDigger funciona como um reconhecedor de padrões altamente treinado sobreposto a um método experimental já poderoso. Ao aprender como sinais entrecruzados genuínos devem parecer em várias dimensões ao mesmo tempo, ele consegue peneirar conjuntos de dados vastos e bagunçados, descartar impostores prováveis e resgatar conexões proteicas reais mas anteriormente ocultas. Embora aplicações em todo o proteoma continuem exigindo grande poder computacional, este trabalho mostra que combinar experimentos de entrecruzamento com aprendizado profundo pode aguçar consideravelmente nossa visão de como as proteínas estão organizadas e com quem se encontram dentro da célula.
Citação: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
Palavras-chave: interações proteicas, espectrometria de massa com entrecruzamento, aprendizado profundo, proteômica, aquisição independente de dados