Clear Sky Science · pt
Uma referência genérica definida por picos consenso para análise de dados de ATAC-seq de célula única
Por que mapear as portas abertas do nosso DNA importa
Cada célula do seu corpo carrega essencialmente o mesmo DNA, mas neurônios, células sanguíneas e células tumorais se comportam de maneiras muito diferentes. Uma razão chave é que apenas certos trechos de DNA estão expostos e “abertos” para uso em cada momento. Novas tecnologias de célula única podem agora medir essa abertura em todo o genoma, mas até agora faltava um mapa de referência comum — algo como um atlas padrão — para comparar resultados entre experimentos e laboratórios. Este estudo constrói esse mapa, chamado cPeaks, e mostra como ele pode afiar nossa visão sobre tipos celulares, desenvolvimento e câncer.
Transformando muitos experimentos em um mapa compartilhado
Os autores começaram reunindo 624 experimentos de alta qualidade que mediram a cromatina acessível — as partes do DNA acessíveis — em mais de 40 órgãos humanos. Em cada experimento, programas de computador já haviam marcado “picos” onde o DNA estava especialmente exposto. Em vez de tratar cada conjunto de dados separadamente, a equipe sobrepôs cuidadosamente todas essas listas de picos ao longo do genoma e mesclou regiões sobrepostas. Em seguida, examinaram com que frequência cada posição minúscula dentro dessas regiões mescladas era chamada de aberta entre os experimentos, transformando cada região em uma forma característica que refletia quão consistentemente aparecia. Quando uma região mesclada continha na verdade vários sítios abertos próximos, eles a dividiram em múltiplas unidades mais simples. Essas unidades — cerca de 1,4 milhão no total — tornaram-se os picos consenso observados, ou cPeaks, um catálogo de referência candidato para a acessibilidade da cromatina humana. 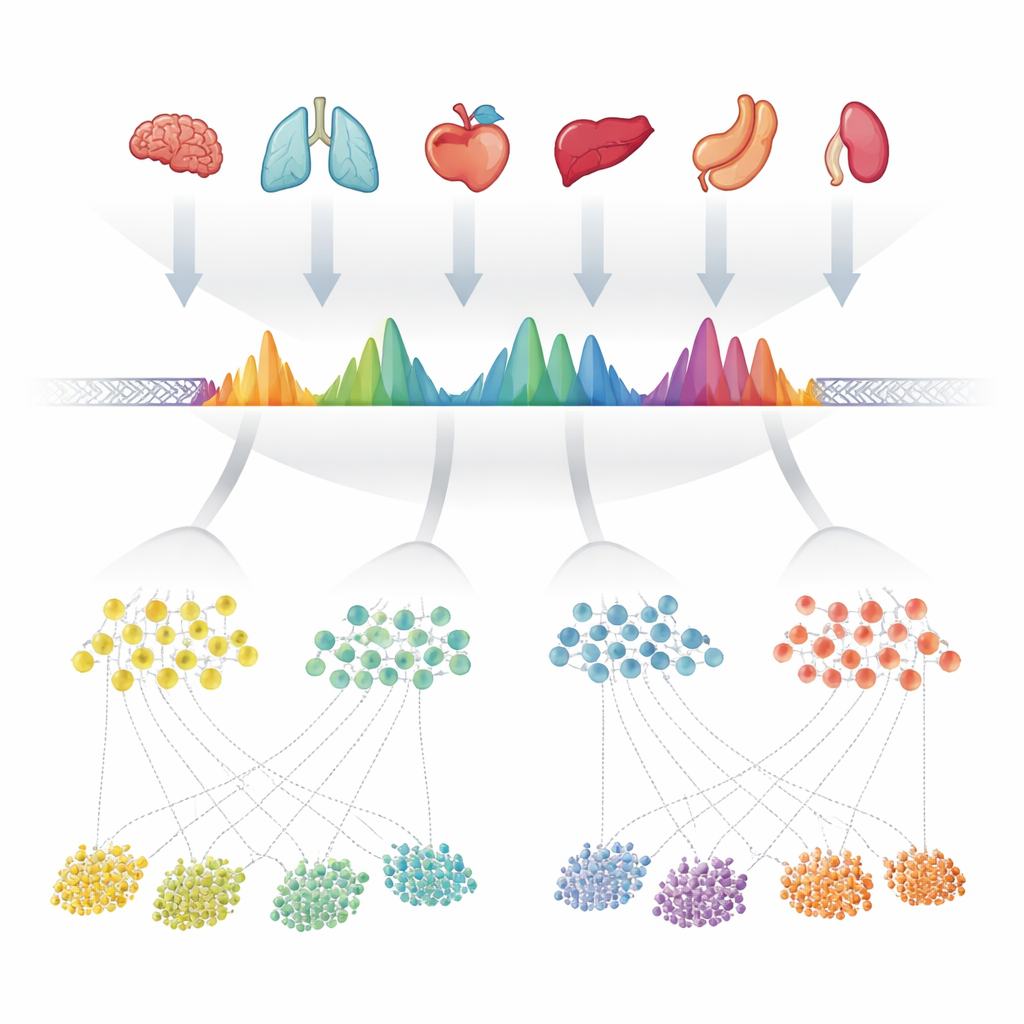
Uma impressão digital estável entre tecidos e tecnologias
Para ser uma referência útil, esses cPeaks devem representar características genuínas e repetíveis do genoma, não peculiaridades de amostras ou de softwares específicos. Os autores testaram isso recriando suas regiões mescladas usando apenas amostras de sangue, apenas tecidos sólidos, bancos de dados públicos separados e até diferentes métodos laboratoriais para sondar o DNA aberto. Em cada caso, os mesmos locais genômicos produziram formas de pico notavelmente semelhantes, e a maioria dos conjuntos de dados de célula única que examinaram sobrepôs mais de 90% de seus próprios picos com o catálogo cPeak. Leitura de muitos órgãos se acumularam precisamente ao redor dos centros dos cPeaks, mostrando que essas regiões capturam de forma confiável onde a cromatina está aberta. Em comparação com conjuntos de referência anteriores baseados em tecnologias relacionadas, os cPeaks cobriram mais do DNA acessível detectado por experimentos ATAC-seq e capturaram quase tanto sinal quanto picos definidos diretamente em cada conjunto — apesar de serem fixos e reutilizáveis.
Ensinando uma rede neural a encontrar regiões ausentes
Mesmo centenas de amostras existentes não conseguem cobrir todo tipo celular possível. Para estender seu mapa a regiões ainda não observadas, a equipe recorreu ao aprendizado profundo. Treinaram uma rede neural convolucional unidimensional em sequências de DNA: exemplos dentro dos cPeaks observados serviram como positivos, enquanto regiões de fundo escolhidas aleatoriamente serviram como negativos. O modelo aprendeu a distinguir esses dois com alta precisão, implicando que os cPeaks carregam padrões de sequência reconhecíveis. Quando os pesquisadores deliberadamente esconderam picos específicos de um tecido por vez, a rede ainda os recuperou apenas pela sequência, incluindo sítios raros e específicos de tecido. Eles então deslizaram uma pequena janela pelo restante do genoma, pontuando cada segmento e adicionando cerca de 280.000 novas regiões de alta pontuação ao catálogo como cPeaks preditos, melhorando particularmente a cobertura em tecidos sub-representados nos dados originais.
Ligando regiões abertas a genes, tipos celulares e células raras
Com uma referência mais rica em mãos, os autores perguntaram o que essas regiões fazem. Muitos cPeaks ficam próximos a sítios de início e término de genes ou se sobrepõem a elementos regulatórios conhecidos, como promotores, enhancers e sítios de ligação para proteínas arquitetônicas como CTCF. Um pequeno subconjunto é acessível em quase todos os conjuntos de dados; esses cPeaks “housekeeping” mais longos tendem a se localizar em regiões de promotores centrais de genes necessários para a manutenção básica da célula. A equipe também classificou os cPeaks pela nitidez e consistência de suas bordas entre as amostras, o que reflete quão precisamente o DNA próximo está empacotado em nucleossomos. Regiões com limites bem definidos são enriquecidas para famílias particulares de fatores de transcrição conhecidas por remodelar a cromatina e conduzir o desenvolvimento. Quando os cPeaks foram usados como conjunto de features para analisar múltiplos conjuntos de dados de célula única, melhoraram a precisão da rotulagem de tipos celulares e foram especialmente úteis para identificar tipos celulares raros e subtipos sutis que conjuntos de picos anteriores ou grades genômicas simples muitas vezes confundiam.
Seguindo o desenvolvimento e o câncer usando uma linguagem comum
O poder de uma referência padrão fica claro ao comparar contextos biológicos muito diferentes. Usando cPeaks, os autores reanalisaram dados de célula única da retina humana em desenvolvimento, grandes atlas de tecidos fetais e adultos, e vários cânceres. Conseguiram reconstruir trajetórias de desenvolvimento e observaram que a fração de cPeaks com limites nítidos e “bem posicionados” tende a aumentar durante estágios transitórios, depois diminuir à medida que as células se estabelecem em identidades estáveis. Um padrão semelhante apareceu ao longo dos estágios tumorais: cânceres intermediários mostraram uma proporção maior dessas regiões estruturadas, sugerindo intenso replanejamento regulatório. Em um tumor ovariano, os cPeaks ajudaram a revelar dois subclones distintos de células cancerígenas com diferenças de número de cópias de DNA, demonstrando como a referência pode expor complexidade oculta na doença.
O que isso significa para a pesquisa genômica futura
Para não especialistas, os cPeaks podem ser pensados como um conjunto padronizado de coordenadas que marcam onde o genoma é mais provavelmente fisicamente aberto e ativo em muitos tipos celulares humanos. Ao alinhar novos experimentos de cromatina de célula única a esse mapa compartilhado, pesquisadores podem comparar resultados entre estudos, identificar com mais facilidade estados celulares raros ou transitórios e começar a construir modelos em larga escala de regulação gênica — assim como catálogos padronizados de genes possibilitaram o surgimento de atlas de RNA de célula única. O catálogo cPeak atual é um rascunho inicial que crescerá à medida que novos dados chegarem, mas já fornece uma linguagem comum para descrever a acessibilidade da cromatina, aproximando-nos de uma visão unificada sobre como o empacotamento do DNA guia o desenvolvimento, a saúde e a doença. 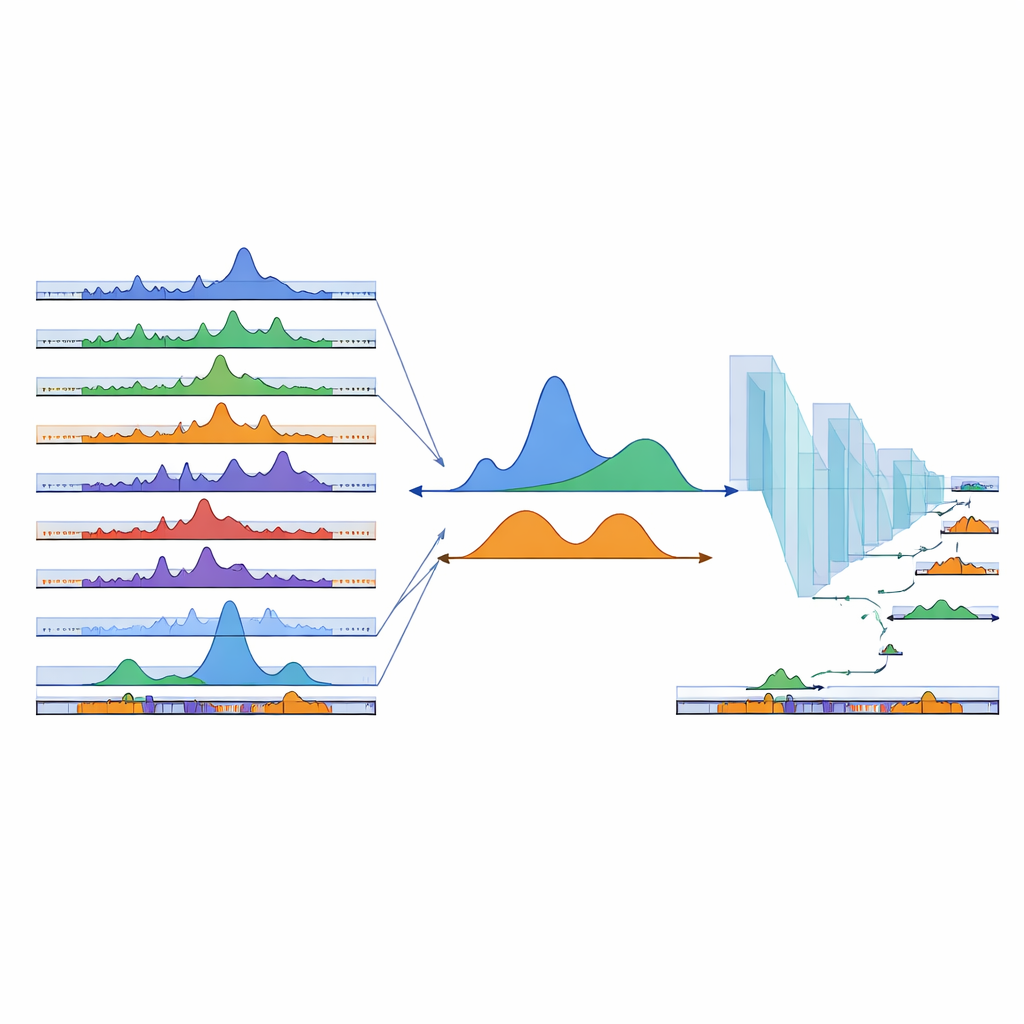
Citação: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Palavras-chave: acessibilidade da cromatina, ATAC-seq de célula única, picos consenso, regulação gênica, genômica por aprendizado profundo