Clear Sky Science · pt
Sequenciamento da metilação e hidroximetilação do DNA em características cromatínicas coocorrentes
Lendo as anotações químicas das nossas células
Cada célula do corpo carrega o mesmo DNA, mas células do cérebro, da pele e células-tronco se comportam de maneiras muito diferentes. Uma razão é que as células escrevem “anotações” químicas no DNA e nas proteínas que o embalam, ajudando a ligar ou desligar genes. Até agora, os cientistas tiveram dificuldade em ler várias dessas anotações juntas no mesmo trecho de DNA, deixando uma lacuna em nossa compreensão de como elas atuam em conjunto. Este estudo apresenta uma nova forma de ler simultaneamente o código genético e marcas químicas-chave, revelando como elas se combinam para controlar interruptores importantes do DNA chamados enhancers.
Por que o DNA precisa de marcas de lápis
O DNA não age sozinho. Ele está enrolado em proteínas chamadas histonas para formar a cromatina, e tanto o DNA quanto as histonas podem ser decorados com pequenos grupos químicos. Duas marcas importantes no DNA são grupos metil e hidroximetil adicionados à letra C (citidina). Essas marcas influenciam o quão compactado o DNA está e se genes próximos estão ativos. Em termos gerais, marcas de metilação estão frequentemente associadas ao silenciamento gênico, enquanto marcas de hidroximetilação tendem a aparecer onde genes estão ativos. Mas o impacto dessas marcas depende do contexto local: exatamente onde elas se situam no genoma e ao lado de quais marcas de histona aparecem.
O problema com mapas separados
Métodos de sequenciamento existentes podem mapear marcas de metilação e hidroximetilação em todo o genoma, e outros métodos mapeiam marcas de histona que sinalizam regiões ativas ou silenciosas. No entanto, isso normalmente é feito em experimentos separados e depois comparado por computador. Isso nos diz quais características tendem a estar na mesma vizinhança, mas não se elas realmente coexistem no mesmo pedaço de DNA dentro de uma única célula. Tentativas anteriores de combinar essas medidas dependiam de tratamentos químicos agressivos que danificavam o DNA e, crucialmente, não conseguiam distinguir de forma confiável metilação de hidroximetilação na mesma leitura. Como resultado, os pesquisadores não tinham uma imagem molecular clara de como combinações de marcas cooperam.
Um novo método de leitura em múltiplas camadas
Os autores desenvolveram um método chamado 6-base-CUT&Tag que pode ler as quatro bases do DNA mais dois estados químicos da citosina—simples, metilada e hidroximetilada—em fragmentos de DNA fisicamente ligados a características cromatínicas selecionadas. Primeiro, usam anticorpos como ganchos moleculares para puxar DNA enrolado em histonas que carregam uma marca específica, por exemplo uma etiqueta de cromatina ativa. Uma enzima projetada então insere adaptadores especiais, transformando cada fragmento de DNA capturado em um pequeno laço que resiste às etapas de limpeza que destroem fragmentos soltos. Um processo químico e enzimático refinado converte os vários estados de citosina em sinais de sequência distintos, que os sequenciadores modernos podem ler. Dessa forma, uma única leitura informa de onde veio o fragmento, qual marca de histona ele carregava e quais citosinas nele estavam metiladas ou hidroximetiladas.
Aproximando-se dos interruptores gênicos
Usando células-tronco embrionárias de camundongo como caso de prova, a equipe aplicou 6-base-CUT&Tag a várias marcas de histona-chave que marcam diferentes tipos de DNA regulatório. Eles focaram em enhancers—trechos de DNA que atuam como interruptores para controlar quando e onde genes são ativados. Enhancers podem estar em estados “ativos”, “preparados” (primed) ou “prontos” (poised), distinguidos por marcas de histona particulares. Os pesquisadores descobriram que enhancers marcados apenas por uma etiqueta de histona chamada H3K4me1 (frequentemente considerada “preparada”) apresentavam os níveis mais altos tanto de metilação quanto de hidroximetilação no DNA, especialmente quando examinados diretamente nos nucleossomos ligados a H3K4me1. Em contraste, enhancers com sinais adicionais de forte atividade ou de repressão exibiam menos dessas marcas de DNA ou mostravam um deslocamento em direção à hidroximetilação, sugerindo um apagamento em andamento das marcas de metilação.
Decodificando estados de enhancer com maior precisão
Porque todos os tipos de enhancer compartilham a marca H3K4me1, a equipe investigou se o padrão detalhado de marcas de DNA especificamente no DNA marcado por H3K4me1 poderia, por si só, distinguir os diferentes estados de enhancer. Eles treinaram um modelo de aprendizado de máquina usando os dados do 6-base-CUT&Tag para classificar enhancers como ativos, preparados ou prontos, com base unicamente na quantidade de metilação e hidroximetilação que carregavam naquela única característica de histona. Esse modelo superou outro modelo idêntico treinado com dados padrão de todo o genoma que não são restritos a nenhuma marca de histona. Em outras palavras, ler marcas de DNA no contexto imediato onde ocorrem dá uma imagem mais nítida do que fazer uma média por todo o DNA da célula.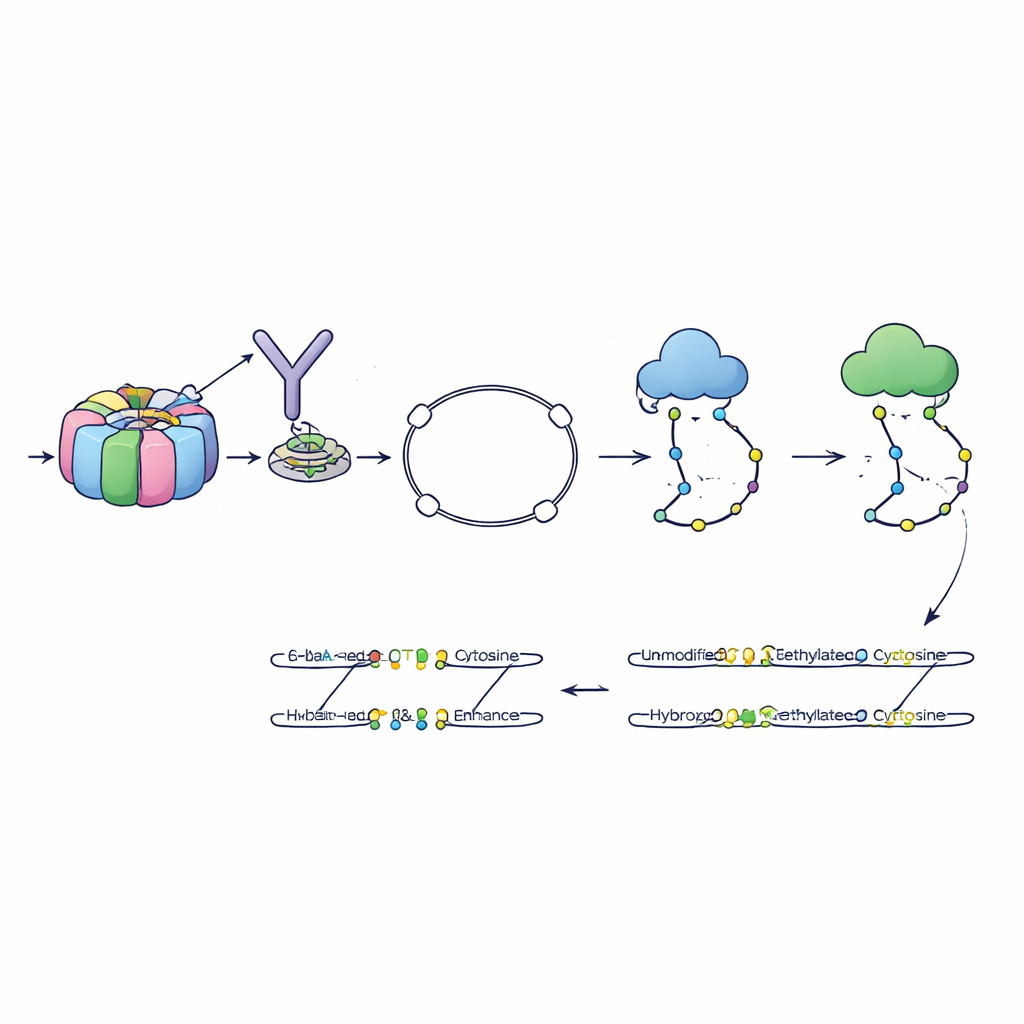
O que isso significa para entender a identidade celular
Para um não especialista, a mensagem chave é que esse método permite aos cientistas ler várias camadas de informação—sequência de DNA, marcas no DNA e marcas de histona—na mesma molécula. Essa visão de alta resolução revela como combinações particulares de etiquetas químicas definem o grau de prontidão dos interruptores gênicos em células-tronco. Como o 6-base-CUT&Tag é mais eficiente e menos danoso do que abordagens anteriores, ele pode revelar padrões sutis que antes estavam ocultos. Com o tempo, essa leitura em múltiplas camadas da cromatina pode ajudar a explicar como as células lembram sua identidade, como mudam durante o desenvolvimento ou em doenças, e como poderíamos direcionar com maior precisão o código regulatório em terapias.
Citação: Araujo Tavares, R.d.C., Dhir, S., He, X. et al. Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features. Nat Commun 17, 2591 (2026). https://doi.org/10.1038/s41467-026-69429-6
Palavras-chave: epigenética, metilação do DNA, cromatina, enhancers, células-tronco