Clear Sky Science · pt
Variantes de perda de função no ativador CAPN1 CD99L2 causam ataxia espástica ligada ao X
Por que isso importa para famílias com problemas de movimento sem explicação
Muitas pessoas vivem anos com dificuldades para caminhar, rigidez muscular ou problemas de equilíbrio e fala sem descobrir a causa real. Este estudo mostra como testes de DNA modernos podem finalmente dar respostas a algumas dessas famílias. Os pesquisadores não só compararam diferentes testes genéticos para distúrbios raros do movimento, como também descobriram uma causa até então desconhecida de uma condição chamada ataxia espástica ligada ao X, apontando para vias biológicas que também podem ser importantes em doenças cerebrais mais comuns.
Encontrando agulhas genéticas no palheiro das doenças raras
Distúrbios raros do movimento, como ataxia (movimentos instáveis) e paraplegia espástica (pernas rígidas e fracas), frequentemente são suspeitos de origem genética, mas para a maioria dos pacientes os testes padrão dão negativo. A equipe acompanhou 2.811 pessoas na Alemanha e em toda a Europa que foram encaminhadas por suspeita de distúrbios raros do movimento ao longo de seis anos. Primeiro, eles analisaram testes direcionados tradicionais que procuram expansões de repetição conhecidas em um pequeno grupo de genes; esses testes forneceram respostas em cerca de 11% dos casos. Em seguida, usaram sequenciamento do exoma, que lê apenas as partes codificadoras de proteínas do genoma, e encontraram explicações genéticas definitivas em cerca de 19% dos pacientes, especialmente entre aqueles com espasticidade.
Indo além dos testes padrão com sequenciamento do genoma completo
Para avançar mais, os cientistas usaram sequenciamento do genoma completo, que lê quase todo o DNA de uma pessoa, incluindo regiões que testes padrão e exomas podem perder. Entre 486 indivíduos submetidos a esse exame mais abrangente, a taxa de diagnóstico aumentou em aproximadamente 7,5 pontos percentuais, sobretudo porque o sequenciamento do genoma identifica melhor alterações complexas, como rearranjos estruturais e expansões de repetição. O estudo também mostrou que informações clínicas registradas com cuidado — em especial descrições específicas dos sintomas, idade mais jovem no momento do teste e a combinação de espasticidade com outros problemas de movimento — ajudaram a prever quem teria maior probabilidade de receber um diagnóstico genético claro.
Descobrindo uma nova causa ligada ao X de ataxia espástica
Mesmo após esses testes extensivos, muitos pacientes permaneceram sem diagnóstico. Os pesquisadores reuniram dados genéticos de mais de 13.000 indivíduos e usaram uma abordagem de “carga gênica”, perguntando quais genes carregavam variantes suspeitas com mais frequência em pacientes do que em controles não afetados. Essa análise apontou não só para genes de doença já conhecidos, como também destacou de forma consistente um gene até então negligenciado no cromossomo X chamado CD99L2. Ao combinar resultados de várias famílias pela Europa, identificaram 25 homens afetados de 20 famílias que carregavam variantes deletérias nesse gene. Esses homens tipicamente desenvolveram problemas para caminhar, rigidez nas pernas, fala arrastada e às vezes dificuldades de equilíbrio na meia‑idade ou mais tarde, enquanto as portadoras femininas eram na maior parte não afetadas — padrões que se encaixam com um distúrbio ligado ao X. As variantes destruíam principalmente a proteína normal ou removiam partes cruciais dela, sugerindo fortemente que a perda de função causa a doença.
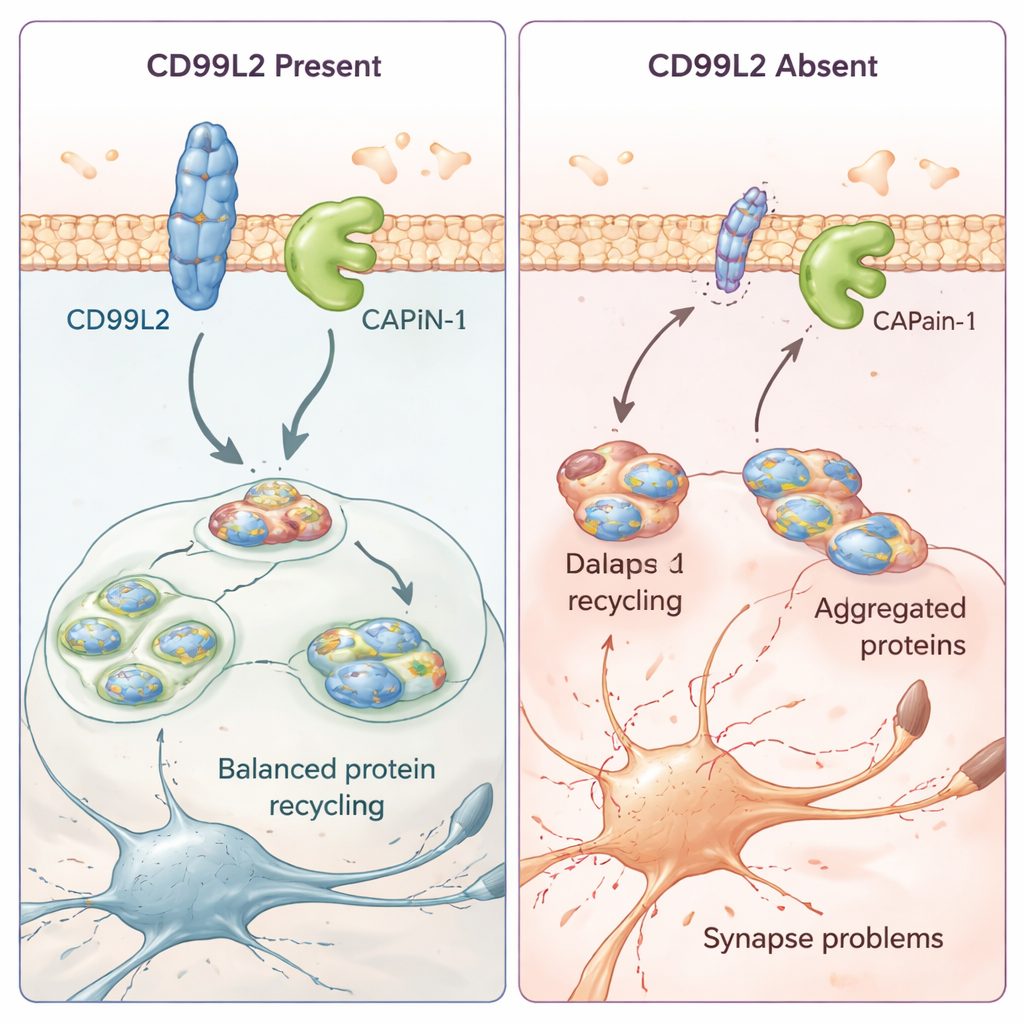
Como uma pequena proteína de membrana ajuda a proteger células cerebrais
Para entender o que o CD99L2 realmente faz nas células, a equipe recorreu a modelos celulares e células da pele derivadas de pacientes. Eles descobriram que a proteína CD99L2 fica na membrana celular e normalmente é marcada com pequenas etiquetas de “ubiquitina” que controlam quanto tempo ela sobrevive antes de ser degradada. O CD99L2 se liga fisicamente à calpaína-1 (CAPN1), uma enzima ativada por cálcio que corta outras proteínas e ajuda a manter as sinapses — os pontos de contato entre neurônios — saudáveis. Quando o CD99L2 está presente e intacto, ele ajuda a ligar e desligar a calpaína-1 de maneira controlada, e então é ele próprio clivado e reciclado. Quando o CD99L2 está ausente ou estruturalmente alterado, a ativação da calpaína-1 fica prejudicada. Em células de pacientes, isso anda de mãos dadas com atividade perturbada de muitos genes relacionados a sinapses e comunicação neuronal, sugerindo que mudanças sutis porém amplas na circuitaria cerebral podem estar por trás dos problemas de movimento graduais.
O que isso significa para pacientes hoje e amanhã
Para famílias com ataxia espástica ou paraplegia espástica sem explicação, este trabalho oferece dois tipos de avanço. Primeiro, mostra que usar o sequenciamento do genoma completo precocemente, junto com descrição clínica cuidadosa, pode aumentar de forma notável as chances de um diagnóstico genético definitivo. Segundo, adiciona o CD99L2 à lista de genes que controlam a atividade da calpaína, uma via já implicada em outras ataxias raras e em doenças comuns como Alzheimer e Parkinson. Em termos práticos, o estudo revela um novo “interruptor” de liga‑desliga que ajuda a manter o equilíbrio da manutenção das células cerebrais; quando esse interruptor se quebra, os neurônios se deterioram lentamente, levando à rigidez e à perda de coordenação. Entender esse interruptor pode, eventualmente, abrir caminho para tratamentos que ajustem a atividade da calpaína e protejam células cerebrais em uma variedade de doenças neurológicas.
Citação: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
Palavras-chave: ataxia espástica, distúrbios raros do movimento, sequenciamento genômico, CD99L2, calpaína-1