Clear Sky Science · pt
Estados excitados ferric-oxyl podem explicar ligações ferro-oxigênio alongadas em intermediários catalíticos de peroxidases heme?
Por que as ligações ferro-oxigênio em enzimas importam
Dentro de nossas células, proteínas especializadas chamadas enzimas usam oxigênio para realizar com segurança reações químicas muito reativas. Entre elas, as peroxidases heme dependem de um par ferro–oxigênio em seu núcleo para decompor peróxido de hidrogênio, uma molécula reativa e potencialmente danosa. Há décadas, cientistas discordam sobre a natureza exata dessa ligação ferro–oxigênio: seria mais parecida com uma dupla ligação rígida ou com uma ligação simples mais frouxa — e o que isso implica para o funcionamento dessas enzimas? Este estudo aborda esse mistério usando métodos ultrarrápidos de raios X e cálculos avançados, revelando que a resposta reside em estados excitados fugazes da própria unidade ferro–oxigênio.
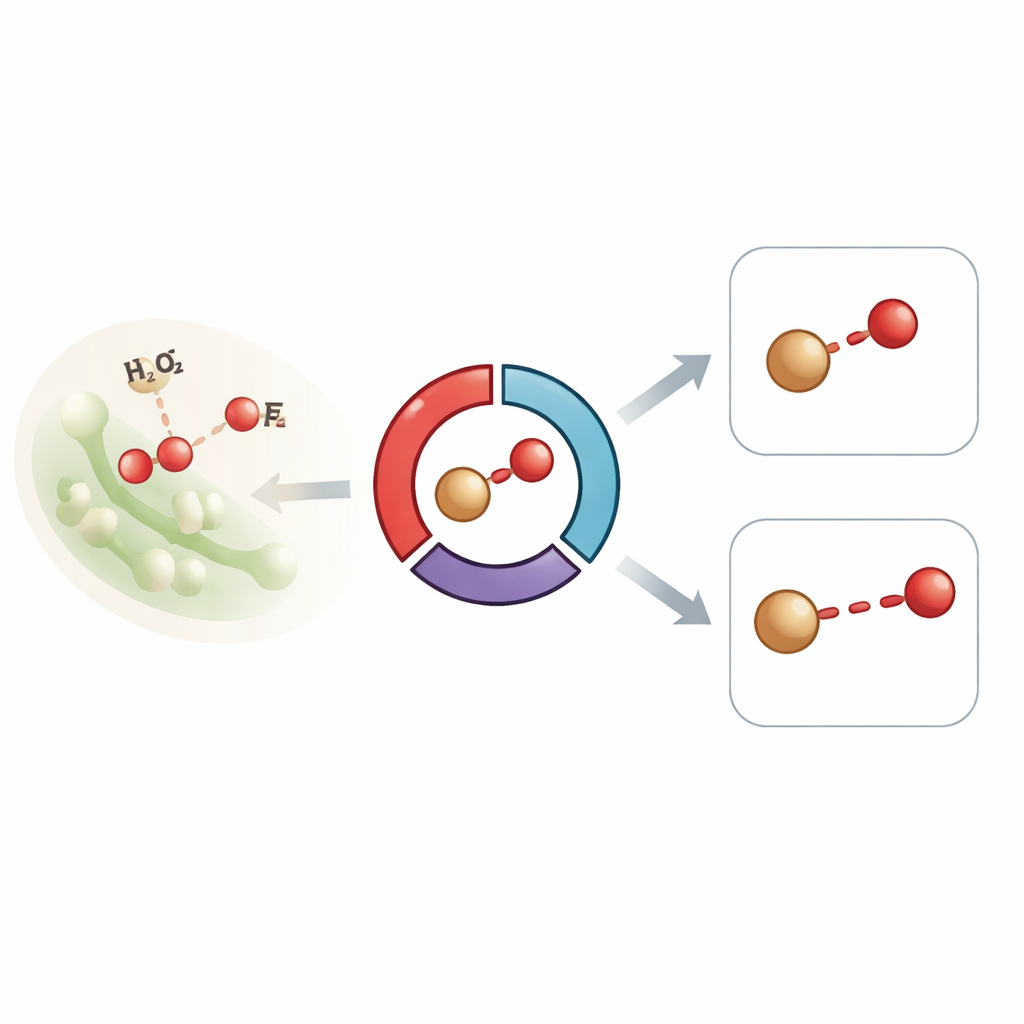
Acompanhando uma enzima em tempo real
Os pesquisadores focaram-se em uma peroxidase bacteriana que descolora corantes, uma enzima heme que normalmente transita por duas formas de alta energia importantes, conhecidas como Composto I e Composto II. Essas formas apresentam ambas um átomo de ferro ligado ao oxigênio e são centrais para como a enzima processa peróxido de hidrogênio e oxida outras moléculas. Experimentos anteriores em enzimas semelhantes registraram comprimentos de ligação ferro–oxigênio surpreendentemente longos, que alguns cientistas interpretaram como evidência de que a unidade ferro–oxigênio altamente reativa teria sido alterada pelos raios X ou teria captado um próton extra, mudando sua natureza. Para evitar tais artefatos, a equipe usou cristalografia seriada por femtossegundo dependente do tempo em um laser de elétrons livres de raios X, capturando difração e sinais de emissão de raios X de milhares de microcristais de proteína em temperatura ambiente, tudo dentro de dezenas de femtossegundos — mais rápido do que o dano pode ocorrer.
Observando a química se desenrolar dentro dos cristais
No arranjo experimental, microcristais de uma versão ligeiramente modificada da enzima foram misturados com peróxido de hidrogênio diretamente sobre uma fita em movimento e então sondados após atrasos que variaram de meio segundo a dezenas de minutos. Pontos de tempo iniciais favorecem a formação do Composto I, enquanto os mais tardios são dominados pelo Composto II. Os dados estruturais mostraram que, em ambos os intermediários, o átomo de ferro fica próximo a um único átomo de oxigênio no bolso do heme, e que regiões de alças protetoras da proteína se deslocam para proteger esse centro altamente oxidante. Importa notar que medições precisas revelaram que o comprimento da ligação ferro–oxigênio permaneceu em torno de 1,83 angstroms em todos os pontos temporais — mais longo do que o esperado para uma espécie ferril clássica (Fe(IV)=O) com dupla ligação e mais próximo de uma ligação simples — ainda que as assinaturas espectrais da emissão de raios X e da absorção óptica indicassem claramente estados de oxidação elevados, compatíveis com os Compostos I e II.
Eliminando explicações simples
Como os experimentos foram realizados com pulsos ultracurtos em temperatura ambiente, os culpados usuais por comprimentos de ligação distorcidos — redução induzida por raios X e artefatos criogênicos — puderam ser amplamente descartados. A equipe também testou se o oxigênio ligado ao ferro teria sido protonado, transformando a dupla ligação em uma ligação simples semelhante a um hidróxido. No entanto, as propriedades ácido–base conhecidas de centros heme semelhantes, juntamente com estudos químicos prévios, argumentam fortemente contra tal protonação nesse tipo de enzima. Os dados espectroscópicos também mostraram que o ferro permaneceu em um estado de alta oxidação e baixo spin após reagir com peróxido de hidrogênio, exatamente como esperado para intermediários ferril autênticos, reforçando a ideia de que a ligação inesperadamente longa deve decorrer de efeitos eletrônicos mais sutis em vez de uma simples mudança de forma química.
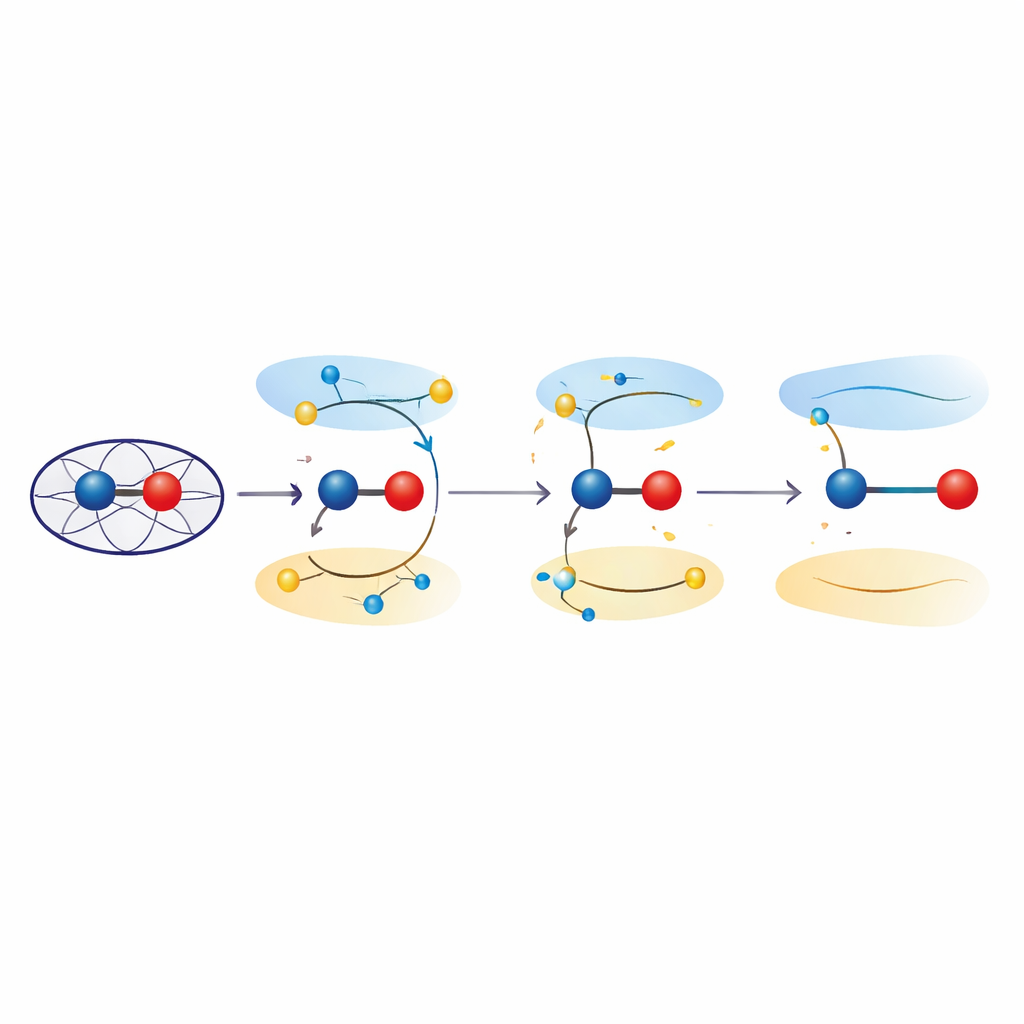
Estados excitados que esticam ligações
Para sondar esses efeitos, os pesquisadores recorreram a cálculos de mecânica quântica tanto em modelos simplificados quanto no ambiente proteico completo. Usando teoria do funcional da densidade dependente do tempo e abordagens combinadas de mecânica quântica/mecânica molecular, eles examinaram como promover elétrons de orbitais ligantes para antiligantes na unidade ferro–oxigênio altera o comprimento preferido da ligação. Esses estados excitados, energeticamente próximos ao estado ferril fundamental, produziram de forma consistente distâncias ferro–oxigênio na faixa de 1,8–1,9 angstrom — em concordância com as observações cristalográficas. A análise da distribuição eletrônica mostrou que, nesses estados, o par ferro–oxigênio deixa de se comportar como uma dupla ligação pura Fe(IV)=O e passa a assumir caráter “ferric–oxyl”, com propriedades semelhantes a Fe(III) ligado a um radical centrado no oxigênio. Refinamentos quânticos das estruturas experimentais confirmaram que descrições em termos de estados excitados ajustam os dados pelo menos tão bem quanto os modelos convencionais no estado fundamental.
O que isso significa para entender o poder das enzimas
Em termos simples, o trabalho sugere que ligações ferro–oxigênio longas observadas nessas peroxidases heme não requerem invocar dano, redução ou prótons ocultos. Em vez disso, elas podem surgir naturalmente quando a unidade ferril acessa brevemente estados excitados de baixa energia que enfraquecem a ligação e conferem caráter ferric–oxyl. Para não especialistas, isso significa que a “parte ativa” de muitas enzimas ativadoras de oxigênio pode ser mais dinâmica e eletronicamente flexível do que se pensava, com mudanças sutis na distribuição eletrônica alterando a força e a reatividade da ligação sem modificar a química global. Reconhecer esses estados excitados pode reformular a maneira como os cientistas interpretam dados estruturais sobre oxidantes biológicos poderosos e pode orientar o desenho de catalisadores artificiais que imitem, ou deliberadamente ajustem, esse delicado equilíbrio eletrônico.
Citação: Williams, L.J., Kamps, J.J., Brânzanic, A.M.V. et al. Can ferric-oxyl excited states explain elongated iron-oxygen bonds in heme peroxidase catalytic intermediates?. Nat Commun 17, 2324 (2026). https://doi.org/10.1038/s41467-026-69192-8
Palavras-chave: peroxidase heme, intermediário ferril, ligação ferro-oxigênio, estados eletrônicos excitados, laser de elétrons livres de raios X