Clear Sky Science · pt
scLong: um modelo de base com bilhões de parâmetros para capturar contexto gênico de longo alcance em transcriptômica de célula única
Ensinando Computadores a Ler a Linguagem Oculta das Células
Cada célula do seu corpo abriga uma cidade movimentada de genes que se ligam e desligam em padrões intrincados. As técnicas modernas de RNA-seq de célula única agora conseguem ouvir cada célula individualmente, mas o resultado é um dilúvio avassalador de números. Este artigo apresenta o scLong, um enorme modelo de inteligência artificial projetado para dar sentido a esses padrões complexos de atividade gênica, incluindo sinais fracos que métodos antigos tendem a ignorar. Seu objetivo é ajudar pesquisadores a entender como as células reagem quando genes são desativados, medicamentos são adicionados ou doenças se instalam.
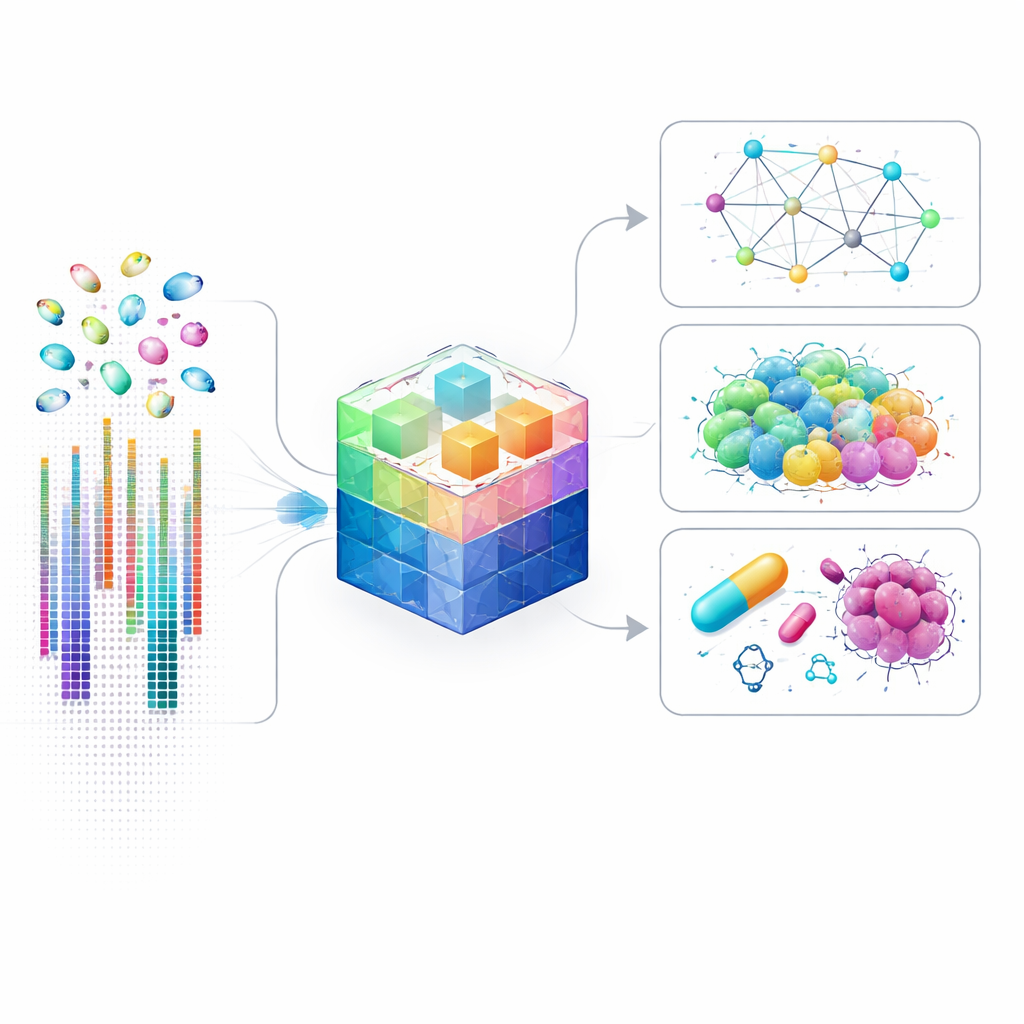
Por Que Mapas Gênicos ao Nível da Célula São Importantes
Estudos gênicos tradicionais costumam misturar milhões de células, fazendo uma média que apaga células raras ou incomuns. As técnicas de célula única mudaram isso ao medir a atividade gênica em cada célula separadamente, revelando tipos celulares ocultos, comunicação sutil entre células e circuitos de controle detalhados que decidem o destino celular. No entanto, analisar esse tipo de dado é extremamente desafiador: cada célula pode ter níveis de atividade medidos para dezenas de milhares de genes, muitos dos quais são mal detectáveis. Modelos de IA existentes simplificam o problema ao focar apenas nos genes mais expressos, o que acelera o cálculo mas perde muitos sinais sutis que podem ser cruciais em doenças, desenvolvimento ou resposta a medicamentos.
Um Novo Modelo que Escuta Cada Gene
O scLong enfrenta esse desafio crescendo em escala em vez de reduzir o problema. É um modelo de base com bilhões de parâmetros treinado em perfis de atividade gênica de cerca de 48 milhões de células humanas de mais de 50 tecidos. Ao contrário de abordagens anteriores que consideram apenas alguns milhares de genes altamente ativos, o scLong leva em conta aproximadamente 28.000 genes de uma vez, incluindo aqueles raramente ou fracamente expressos. Ele combina dois tipos de informação para cada gene: quão ativo ele está numa célula dada e o que já se sabe sobre sua função a partir da Gene Ontology, um grande catálogo curado por especialistas sobre papéis e relações gênicas. Uma rede especializada operando sobre um grafo de conexões gênicas destila esse conhecimento prévio em representações compactas que o modelo pode usar junto aos valores brutos de expressão.
Como o Modelo Equilibra Potência e Eficiência
Analisar cada gene em detalhe é computacionalmente caro, por isso o scLong usa um desenho inteligente de duas pistas. Dentro de cada célula, os genes são ordenados pela intensidade de sua expressão. Os genes mais ativos, que frequentemente carregam o sinal biológico principal, são processados por um módulo de atenção maior e mais poderoso. Os genes mais silenciosos, incluindo medições baixas e até zeros, passam por um módulo menor e mais leve. Depois, todos os genes são reunidos e passam por outra camada de atenção que permite que cada gene influencie todos os outros. Esse desenho permite que o modelo mantenha representações mais baratas mas ainda significativas para sinais fracos, enquanto reserva mais capacidade para os mais fortes. Durante o pré-treinamento, o sistema esconde repetidamente um subconjunto dos valores de atividade gênica e aprende a reconstruí-los a partir do contexto circundante, forçando-o a descobrir os padrões que conectam os genes entre si.
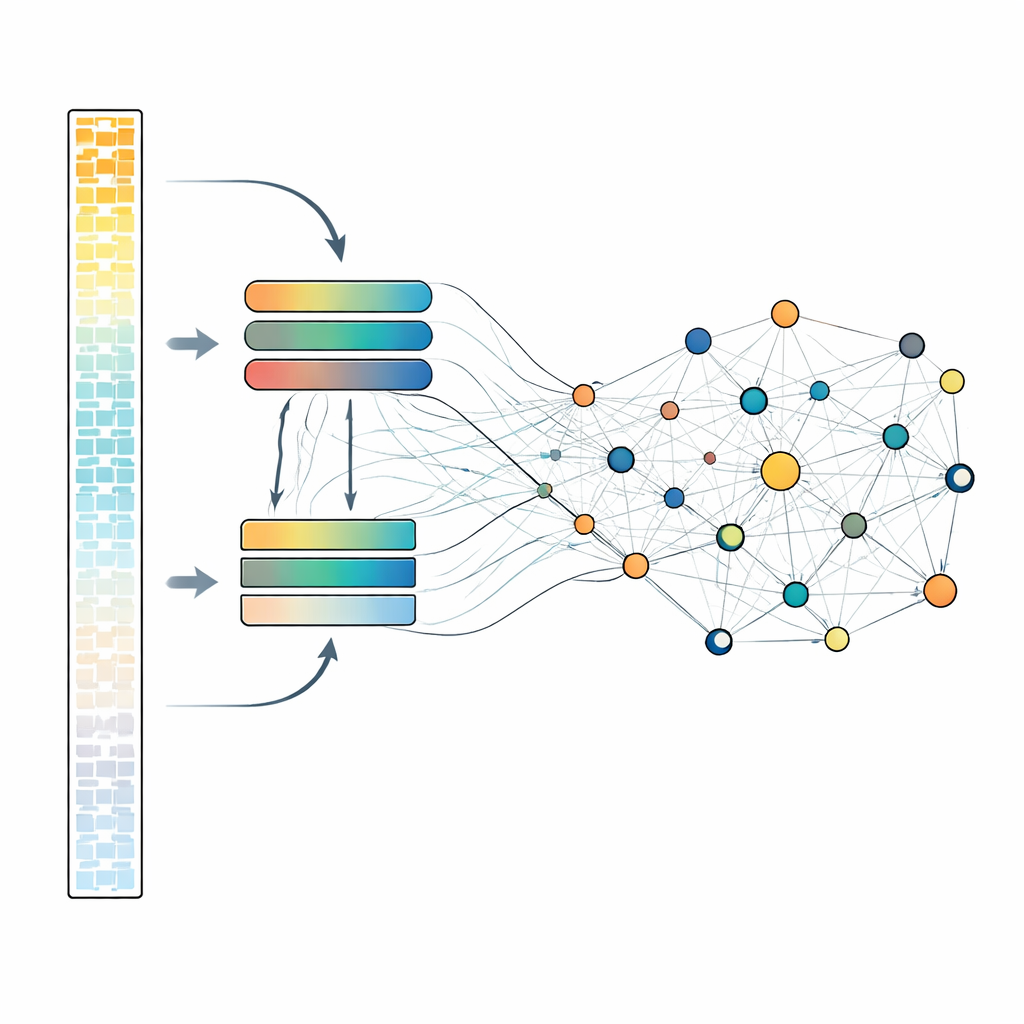
Aplicando o Modelo a Problemas Reais
Uma vez treinado, o scLong pode ser adaptado a uma ampla gama de questões biológicas. Os autores mostram que ele prevê como a atividade gênica mudará quando genes específicos são desativados ou alterados, incluindo combinações de dois genes que podem atuar em conjunto. Também antecipa como as células respondem quando expostas a diferentes compostos químicos, o que é importante para descoberta de fármacos e testes de segurança. Em estudos de câncer, o scLong ajuda a prever como linhas celulares tumorais reagirão a fármacos isolados e a pares de fármacos que podem funcionar melhor em combinação, frequentemente superando tanto modelos especializados quanto outros grandes modelos de base. Para além da predição, o scLong pode inferir redes de relações regulatórias entre genes e ajudar a corrigir distorções técnicas que surgem quando dados são coletados em laboratórios ou máquinas diferentes.
O Que Isso Significa para a Medicina e a Pesquisa Futuras
Em termos simples, o scLong oferece aos cientistas um mapa de alta resolução e sensível ao contexto da atividade gênica dentro de células individuais, um mapa que não descarta genes silenciosos ou raramente usados. Ao aprender com milhões de células e incorporar conhecimento biológico existente, ele fornece estimativas mais precisas sobre como as células reagirão quando genes são perturbados, quando novos medicamentos são introduzidos ou quando processos de doença se desenrolam. Isso pode acelerar a busca por novas terapias, orientar escolhas de tratamento mais personalizadas e aprimorar nossa compreensão de como redes gênicas complexas controlam a saúde e a doença. Embora o modelo seja grande e exigente em termos computacionais, ele aponta para um futuro em que sistemas de IA poderosos e multiuso servem como companheiros versáteis para explorar os mecanismos ocultos de nossas células.
Citação: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Palavras-chave: transcriptômica de célula única, modelos de base, regulação gênica, predição de resposta a fármacos, expressão gênica