Clear Sky Science · pt
Revelando a diversidade de PAMs do Cas9 por mineração metagenômica e aprendizado de máquina
Por que isso importa para a edição gênica futura
O CRISPR tornou-se um símbolo da edição gênica moderna, mas uma regra silenciosa ainda limita o que ele pode fazer: cada corte no DNA deve estar ao lado de uma curta sequência “autorizadora”. Esses padrões curtos, chamados PAMs, determinam onde a popular enzima Cas9 pode ou não atuar. Este estudo mostra como vasculhar enormes quantidades de DNA microbiano, combinado com aprendizado de máquina avançado, pode revelar uma imensa variedade oculta dessas sequências autorizadoras. Esse novo mapa pode abrir muito mais pontos no genoma humano para terapias precisas e mais seguras.
Regras ocultas que guiam os cortes do CRISPR
Cas9 e enzimas relacionadas fazem parte de um sistema imune natural encontrado em bactérias e arqueias. Para evitar cortar seu próprio DNA, esses microrganismos fazem com que as proteínas Cas procurem um PAM—um trecho muito curto de letras—ao lado do sítio alvo. Somente quando esse PAM está presente a Cas9 desenrola o DNA e permite que seu RNA-guia verifique a correspondência, desencadeando um corte se tudo coincidir. O problema para a medicina é que ferramentas comuns de laboratório, como a Cas9 padrão de Streptococcus pyogenes, reconhecem apenas padrões estreitos de PAM. Se uma mutação causadora de doença não tiver a sequência adequada nas proximidades, as ferramentas atuais simplesmente não podem alcançá-la sem sacrificar a precisão.
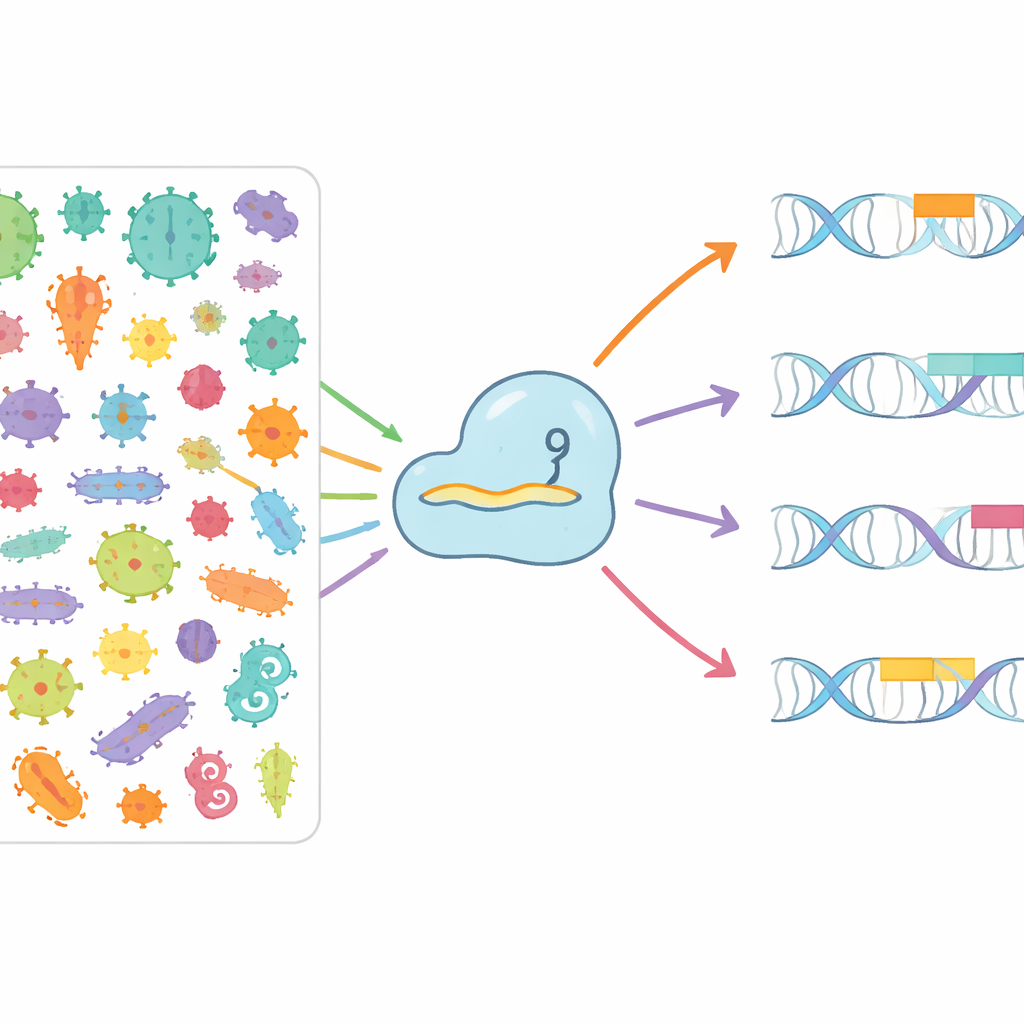
Mineração do mundo microbiano em busca de novas opções
Os autores propuseram-se a mapear sistematicamente como diferentes proteínas Cas9 reconhecem diferentes PAMs na natureza. Eles vasculharam mais de 3,8 milhões de genomas bacterianos e arqueanos e mais de 7,4 milhões de sequências virais e de plasmídeos que infectam ou se movem entre microrganismos. Ao identificar arranjos CRISPR, vinculá-los a genes Cas9 próximos e então emparelhar os “espaçadores” memorizados contra vírus e plasmídeos invasores, puderam ver quais padrões curtos de DNA tendiam a flanquear alvos reais. A partir disso, construíram o CRISPR-PAMdb, um catálogo público contendo 8003 grupos de Cas9, cada um emparelhado com um perfil consensual de PAM, e os organizaram em uma árvore evolutiva que destaca como enzimas Cas9 intimamente relacionadas tendem a compartilhar preferências de PAM semelhantes, ao mesmo tempo em que exibem uma diversidade geral notável.
Quando faltam dados, deixe o modelo aprender
Mesmo com essa pesquisa enorme, a maioria das proteínas Cas9 encontradas não tinha alvos virais suficientes correspondentes para ler diretamente um PAM. Para preencher as lacunas, a equipe construiu um modelo de aprendizado de máquina chamado CICERO. O CICERO usa um poderoso “modelo de linguagem” de proteínas que aprendeu padrões gerais de sequências de aminoácidos e o ajusta para prever, para qualquer proteína Cas9 dada, quão provável é cada letra de DNA aparecer em cada uma de dez posições no PAM. O modelo foi treinado nos perfis de PAM do CRISPR-PAMdb e então testado tanto por validação cruzada quanto em 79 enzimas Cas9 cujos PAMs foram medidos experimentalmente, alcançando forte concordância entre previsão e realidade.
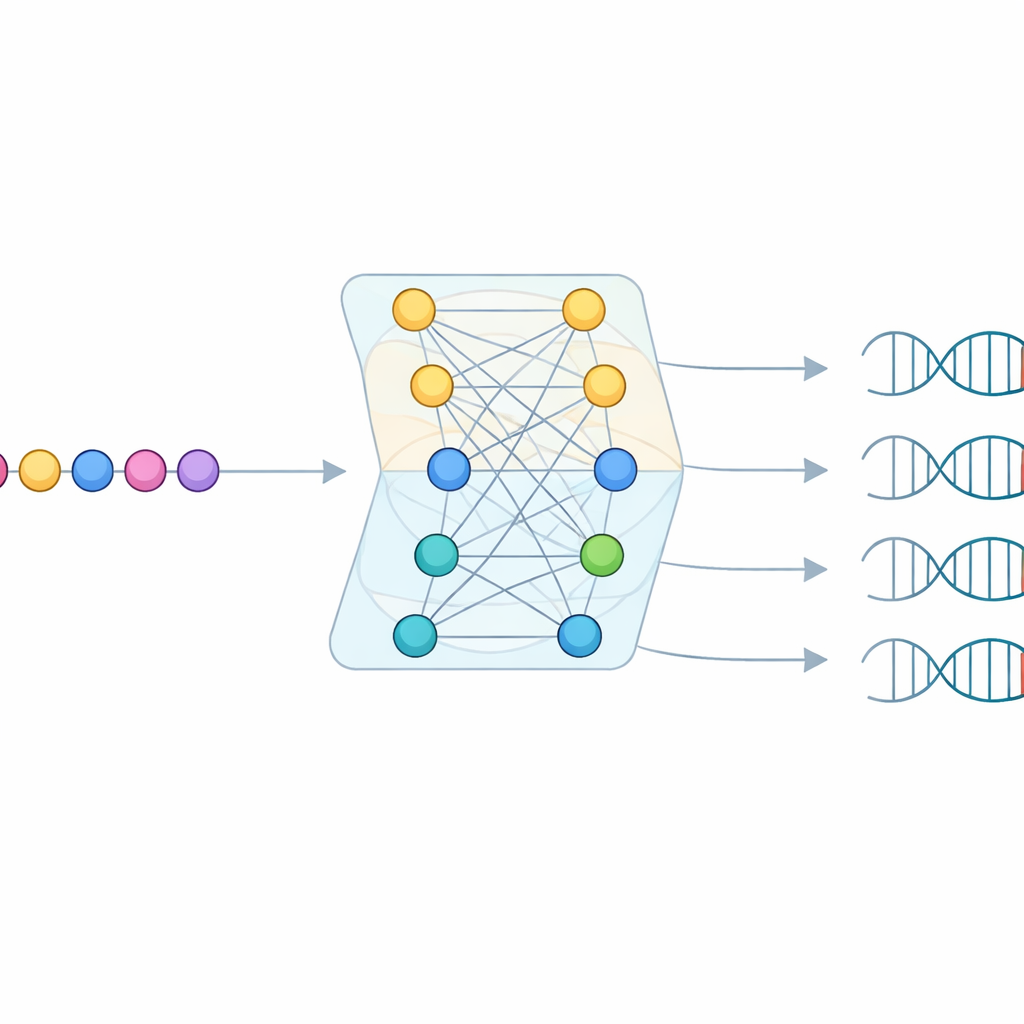
Saber quão confiável é a previsão
Uma característica-chave do CICERO é que ele não apenas adivinha um PAM—ele também estima o quão confiável é cada palpite. Depois de aprender a prever padrões de PAM, os pesquisadores treinaram uma segunda rede leve que recebe a mesma sequência de Cas9 e aprende a prever quão precisa será a previsão de PAM. Pontuações de confiança mais altas se correlacionaram fortemente com maior precisão no mundo real. Usando esse filtro de confiança, a equipe estendeu anotações de PAM para mais de 50.000 proteínas Cas9 adicionais, com mais de 17.000 previsões classificadas como alta confiança. Isso amplia muito o menu de variantes Cas9 com regras de direcionamento razoavelmente bem compreendidas.
O que isso significa para tratar doenças genéticas
Para mostrar por que esses novos recursos importam, os autores examinaram dezenas de milhares de mutações de uma única letra relacionadas a doenças no banco de dados ClinVar que, em princípio, poderiam ser corrigidas usando editores de base—ferramentas que mudam uma letra do DNA sem cortar ambas as fitas. Eles descobriram que a enzima Cas9 padrão pode acessar apenas cerca de metade desses sítios por causa de suas demandas estritas por PAM. Quando permitiram parentes da Cas9 do CRISPR-PAMdb e previsões de alta confiança do CICERO que reconhecem um conjunto mais amplo, mas ainda específico, de sequências próximas, quase todas essas mutações se tornaram teoricamente alcançáveis sem relaxar o direcionamento a ponto de perder precisão.
Uma caixa de ferramentas maior para cirurgia precisa do DNA
Em termos simples, este trabalho constrói duas coisas: um mapa público gigante ligando milhares de proteínas Cas9 naturais aos curtos padrões de DNA que preferem, e um guia de IA que pode prever essas preferências para muitas outras enzimas apenas a partir de suas sequências. Juntos, eles transformam o mundo microbiano em uma rica biblioteca de peças para editores gênicos futuros. À medida que pesquisadores refinarem e testarem essas variantes Cas9 no laboratório, clínicos podem ganhar ferramentas mais seguras e versáteis capazes de alcançar mutações causadoras de doenças que antes estavam fora do alcance, aproximando a cirurgia genômica verdadeiramente precisa da realidade.
Citação: Fang, T., Bogensperger, L., Feer, L. et al. Uncovering Cas9 PAM diversity through metagenomic mining and machine learning. Nat Commun 17, 2510 (2026). https://doi.org/10.1038/s41467-026-69098-5
Palavras-chave: CRISPR-Cas9, diversidade de PAM, metagenômica, aprendizado de máquina, edição genômica