Clear Sky Science · pt
Predição de similaridade química entre espectros de massa em modos de ionização cruzada na metabolômica
Por que conectar pontos químicos importa
Cada gole de café, respiração de ar ou dose de medicamento deixa minúsculos vestígios químicos em nossos corpos. Instrumentos modernos conseguem detectar milhares dessas moléculas ao mesmo tempo, mas transformar esses sinais em compreensão biológica continua surpreendentemente difícil. Este estudo apresenta o MS2DeepScore 2.0, uma ferramenta de aprendizado de máquina que ajuda cientistas a ver como essas moléculas estão relacionadas, mesmo quando os sinais são registrados de maneiras muito diferentes. Ao fazer isso, promete interpretações mais rápidas e mais completas de misturas químicas complexas em medicina, nutrição e pesquisa ambiental.
Duas maneiras de observar a mesma molécula
A espectrometria de massas é uma técnica-central que pesa e fragmenta moléculas para revelar sua identidade. Em experimentos rotineiros, os cientistas frequentemente medem a mesma amostra em dois modos: um que favorece moléculas carregadas positivamente e outro que favorece as carregadas negativamente. Cada modo produz seu próprio “código de barras” característico de fragmentos. Mesmo quando ambas as medições provêm da mesma molécula, os padrões resultantes podem parecer tão diferentes que métodos tradicionais de comparação falham. Como resultado, os pesquisadores geralmente analisam os dois modos separadamente, constroem dois mapas desconectados da amostra e correm o risco de perder relações químicas importantes.
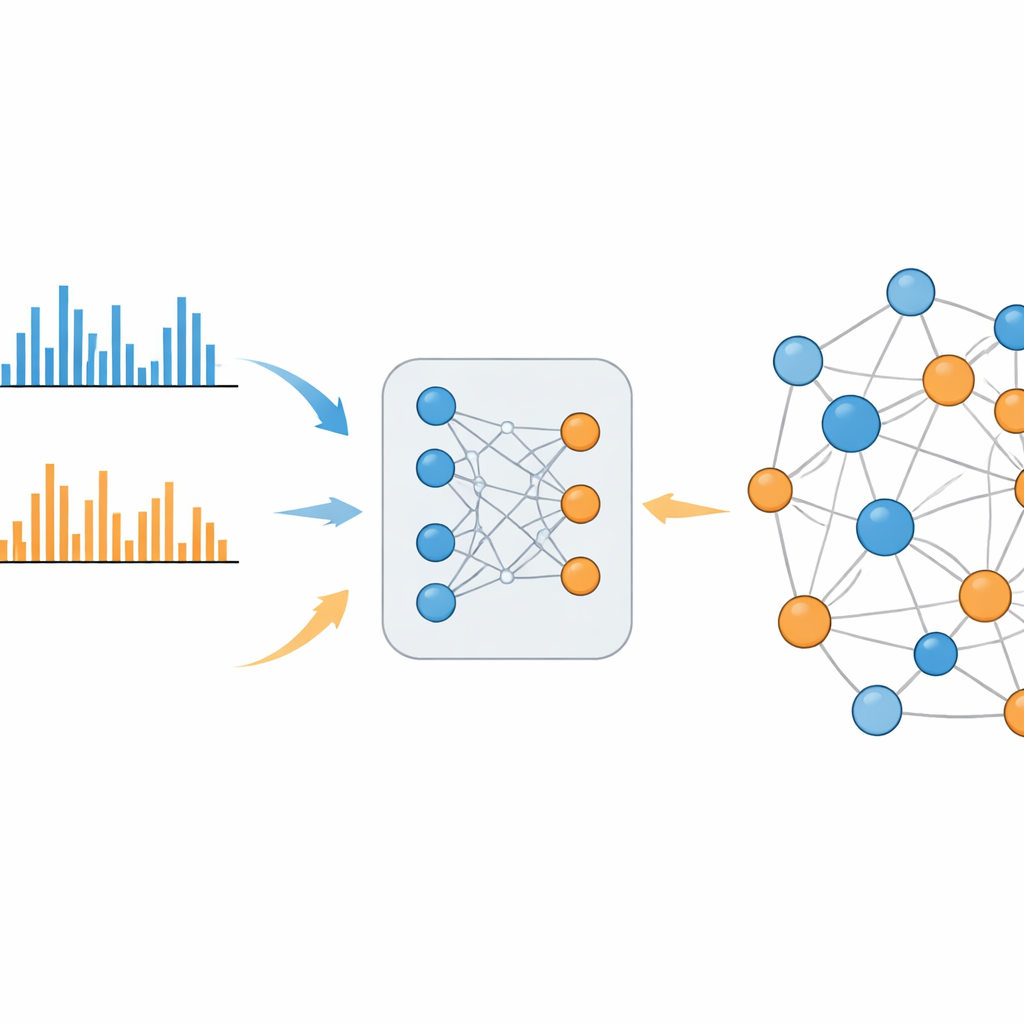
Um sistema de aprendizado que preenche a lacuna
O MS2DeepScore 2.0 enfrenta essa divisão aprendendo similaridade química diretamente a partir de grandes bibliotecas de espectros conhecidos. O modelo é construído sobre um desenho de rede neural gêmea que converte cada padrão de fragmentação em uma impressão digital de 500 números, chamada embedding. Durante o treinamento, o sistema vê centenas de milhares de exemplos dos modos positivo e negativo, junto com o quanto as moléculas subjacentes realmente são semelhantes. Ele se ajusta para que espectros de moléculas relacionadas acabem com embeddings semelhantes, quer tenham sido medidos no mesmo modo ou em modos opostos. A nova versão vai além de sua predecessora ao incluir informações adicionais, como a massa da molécula original e qual modo de ionização foi usado, e ao empregar um esquema de amostragem cuidadosamente balanceado para que relações químicas raras, porém informativas, não sejam abafadas pelas comuns e pouco informativas.
De sinais dispersos a mapas unificados
Depois de treinado, o MS2DeepScore 2.0 pode estimar o quão quimicamente semelhantes dois espectros são, incluindo pares positivo versus negativo. Os autores mostram que essas predições se correlacionam bem com medidas estabelecidas de similaridade estrutural, não apenas dentro de cada modo, mas também entre modos. Usando dados reais de urina humana, plasma sanguíneo humano e uma planta comestível silvestre, eles constroem “redes moleculares” nas quais cada espectro é um nó e alta similaridade prevista cria uma conexão. Diferente de abordagens antigas, essas redes misturam naturalmente dados dos modos positivo e negativo em mapas únicos e coerentes. Agrupamentos curados por especialistas incluem, por exemplo, conjuntos de moléculas relacionadas à cafeína na urina que estão ligadas através dos modos de ionização e correspondem a vias metabólicas conhecidas.
Ver a paisagem química de relance
Redes moleculares são poderosas, mas podem se emaranhar se muitas conexões fracas forem incluídas. Para evitar isso, os autores usam os embeddings do MS2DeepScore diretamente como coordenadas em uma disposição bidimensional criada com uma técnica chamada UMAP. Cada ponto nesse mapa representa um espectro, e pontos próximos correspondem a moléculas que o modelo considera quimicamente semelhantes. Espectros de modos positivo e negativo da mesma substância, que parecem muito diferentes a olho nu, frequentemente aparecem lado a lado nesse espaço de embedding. A equipe também treina um modelo adicional que inspeciona cada embedding e estima quão confiável ele é, sinalizando espectros que são ruidosos, incompletos ou diferentes de tudo visto durante o treinamento. Remover esses pontos de baixa qualidade melhora a precisão geral e torna as visualizações mais confiáveis.
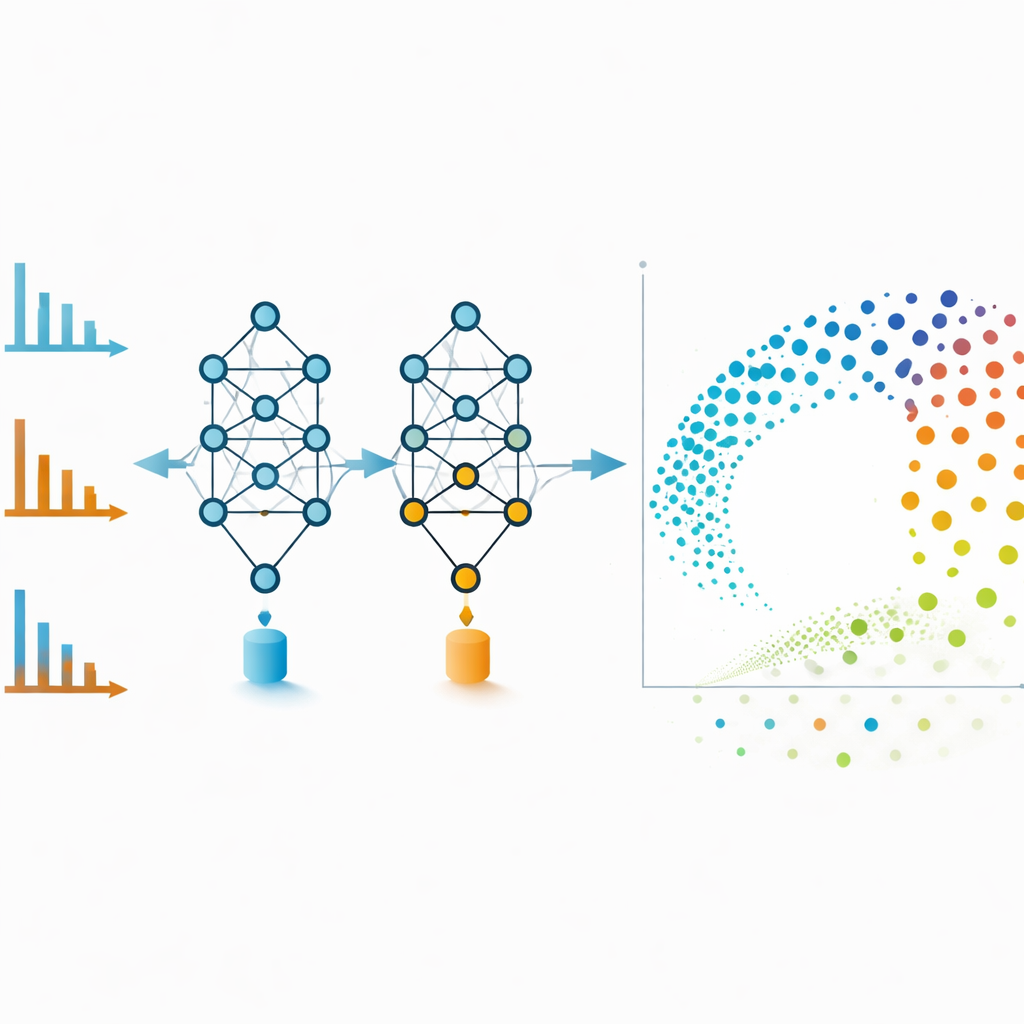
Levando ferramentas avançadas a laboratórios do dia a dia
Para garantir que essa tecnologia seja utilizável além de especialistas em programação, os autores integraram o MS2DeepScore 2.0 a softwares populares e gratuitos de espectrometria de massas. Com essa integração, pesquisadores podem detectar features, construir redes moleculares que ignoram as fronteiras de modo de ionização e explorar o espaço químico resultante por meio de painéis interativos. O código, modelos treinados e conjuntos de dados de exemplo são compartilhados abertamente, e o sistema pode ser re-treinado ou ajustado para classes químicas especializadas.
O que isso significa para descobertas futuras
Para não-especialistas, a mensagem principal é que o MS2DeepScore 2.0 ajuda a transformar medições fragmentadas e dependentes de modo em uma única imagem mais compreensível das moléculas presentes em uma amostra. Ao ligar de forma confiável sinais que antes viviam em mundos analíticos separados, o método permite que cientistas usem bibliotecas de referência muito maiores, comparem amostras de forma mais completa e concentrem sua atenção em agrupamentos significativos de compostos relacionados. Essa interconexão de dados deve acelerar a identificação de biomarcadores, nutrientes, produtos naturais e poluentes, aprofundando, em última instância, nossa compreensão de como a química molda a saúde e o meio ambiente.
Citação: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Palavras-chave: metabolômica, espectrometria de massas, aprendizado de máquina, rede molecular, similaridade química