Clear Sky Science · pt
Avaliação de tecnologias de atlas de ATAC‑seq single‑cell usando modelagem sequência‑para‑função
Lendo o manual de instruções da célula
Todas as células do seu corpo leem o mesmo DNA, porém células cerebrais, musculares e do sistema imune se comportam de formas muito diferentes. Este artigo aborda um enigma central por trás dessa diversidade: como trechos curtos de DNA chamados enhancers atuam como interruptores para ligar e desligar genes em tipos celulares específicos. Os autores mostram que novas tecnologias de laboratório, mais baratas, podem gerar os conjuntos de dados massivos necessários para treinar modelos modernos de deep learning que leem sequências de DNA e predizem quais enhancers estão ativos em quais células, aproximando‑nos de decodificar a “gramática” regulatória do genoma.
Fazendo mapas do DNA acessível em células individuais
Enhancers geralmente ficam em trechos de DNA mais abertos e acessíveis, o que facilita a ligação de proteínas regulatórias. Uma técnica chamada ATAC‑seq single‑cell mede quais partes do genoma estão abertas em milhares a centenas de milhares de células individuais ao mesmo tempo, criando um “atlas” do DNA acessível através de muitos tipos celulares. Esses atlas são combustível ideal para modelos de deep learning que usam a sequência bruta de DNA como entrada e aprendem a predizer com que intensidade cada pequena região atua como enhancer em cada tipo celular. Até agora, porém, a maioria desses atlas dependia de instrumentos comerciais caros, levantando a questão de se métodos open‑source e de baixo custo podem fornecer dados de treinamento de valor equivalente para esses modelos.
Uma alternativa open‑source às plataformas comerciais
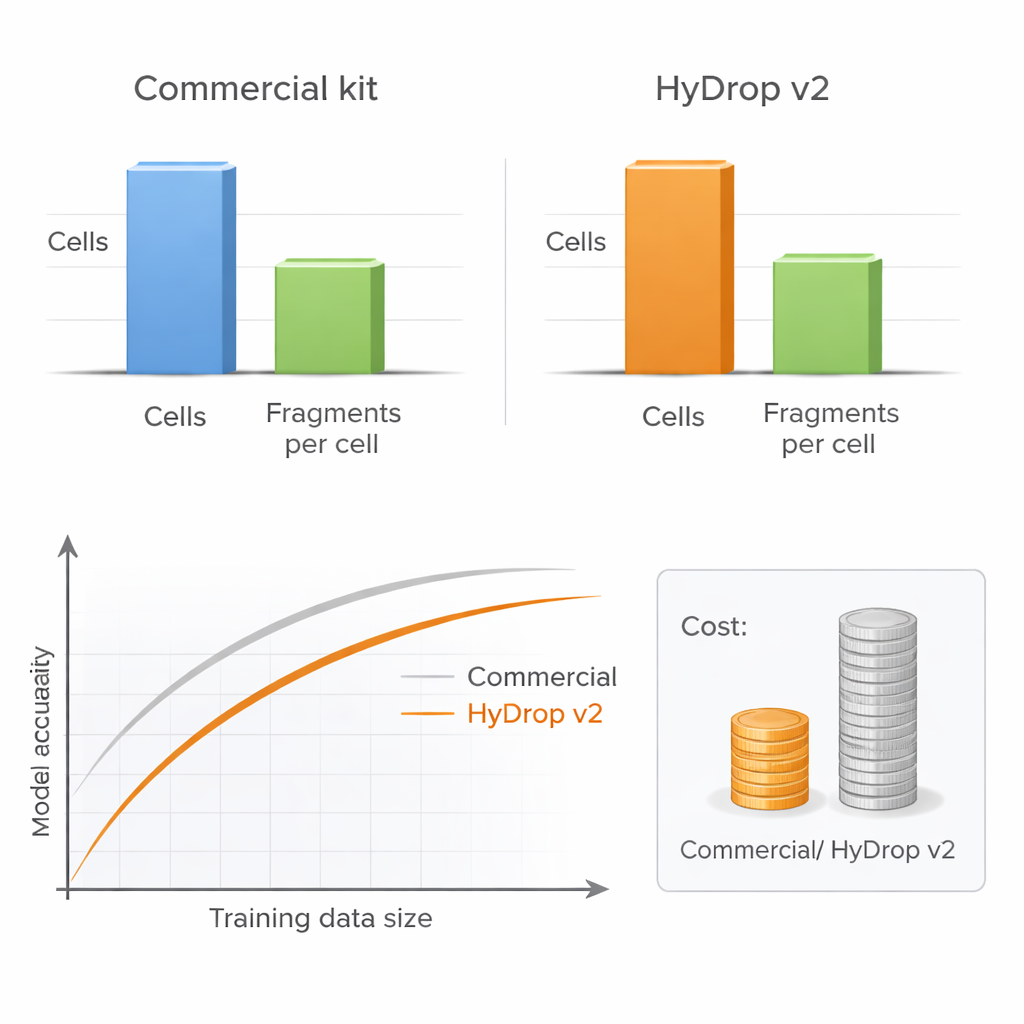
Os autores apresentam o HyDrop v2, um método droplet‑based aprimorado para ATAC‑seq single‑cell que usa bolinhas de hidrogel personalizadas para barcodificar células individuais. Eles comparam o HyDrop v2 com um kit comercial amplamente utilizado, construindo grandes atlas a partir de dois sistemas muito diferentes: o córtex motor de camundongo adulto e embriões tardios da mosca‑da‑fruta. O HyDrop v2 gera qualidade de dados comparável—recuperando os mesmos tipos celulares principais e conjuntos muito semelhantes de regiões de DNA acessíveis—enquanto custa cerca de quatorze vezes menos por amostra de cérebro de camundongo. Importante, os dados de experimentos com HyDrop v2 se integram bem com dados comerciais, o que significa que pesquisadores podem combinar plataformas ao construir atlas muito grandes.
Treinando modelos de deep learning para ler a lógica dos enhancers
Para testar se dados mais baratos são suficientes para modelagem avançada, a equipe treina modelos sequência‑para‑função de deep learning em atlas comerciais ou HyDrop v2. Esses modelos aprendem diretamente a partir da sequência de DNA a predizer quão acessível cada região é em cada tipo celular, e podem destacar padrões curtos de sequência que provavelmente correspondem a sítios de ligação para proteínas regulatórias específicas. No córtex de camundongo, modelos treinados com dados do HyDrop v2 igualam os modelos baseados em dados comerciais em precisão geral e na capacidade de recuperar conhecidos “interruptores” enhancers que foram previamente validados em animais vivos. No embrião da mosca, ambas as plataformas suportam modelos que podem escavar regiões de 2.000 pares de bases e identificar os segmentos centrais de ~500 pares de bases que realmente impulsionam a atividade enhancer específica de tecido, como regiões que controlam a expressão gênica em neuroblastos ou músculos.
Mais células podem superar maior profundidade
Uma questão prática chave para qualquer laboratório é se deve sequenciar cada célula muito profundamente ou perfilar mais células com menor profundidade. Variando sistematicamente o número de células e o número de fragmentos de DNA por célula, os autores mostram que o desempenho dos modelos quase não sofre quando a profundidade de sequenciamento é reduzida a um nível moderado, desde que um número suficiente de células seja incluído. Em contraste, reduzir o número de células prejudica claramente a precisão do modelo, especialmente ao medir desempenho através de muitos tipos celulares ao mesmo tempo. Como o HyDrop v2 é muito mais barato por célula, os pesquisadores podem adicionar facilmente dezenas de milhares de células extras, recuperando ou até superando o desempenho dos modelos baseados em plataformas comerciais a uma fração do custo.
Vendo pegadas de proteínas no DNA
O estudo também examina se diferentes plataformas de laboratório introduzem vieses sutis em como a enzima do ATAC‑seq corta o DNA, o que poderia induzir modelos a erro ao inferir onde as proteínas se localizam no genoma. Usando uma ferramenta neural separada que corrige preferências enzimáticas, os autores mostram que HyDrop v2 e kits comerciais produzem padrões quase idênticos de atividade enzimática em células de camundongo e mosca. Após correção, ambos os conjuntos de dados revelam “pegadas” em escala fina onde proteínas regulatórias e nucleossomos parecem proteger o DNA do corte, e essas pegadas coincidem com os padrões de sequência destacados pelos modelos sequência‑para‑função. Esse acordo sugere que plataformas open‑source e comerciais são igualmente adequadas para estudos detalhados de como proteínas interagem com o DNA.
Por que isso importa para decodificar o genoma
Para não‑especialistas, a mensagem principal é que agora podemos construir mapas muito grandes e acessíveis de como o DNA é usado em células individuais, e treinar poderosos modelos de deep learning com esses mapas sem depender exclusivamente de hardware proprietário caro. O HyDrop v2 fornece dados que suportam predição de enhancers, interpretação de padrões de sequência e pegadas de ligação de proteínas em pé de igualdade com métodos comerciais de ponta, desde que sejam perfis um número suficiente de células. Isso abre a porta para construir atlas abrangentes de elementos regulatórios em organismos, em saúde e doença, acelerando esforços para ler as instruções regulatórias do genoma e projetar novos interruptores genéticos precisamente dirigidos para pesquisa e futuras terapias.
Citação: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Palavras-chave: ATAC‑seq single‑cell, enhancers, modelos de deep learning, regulação gênica, genômica open‑source