Clear Sky Science · pt
Previsão confiável de números da Comissão de Enzimas usando um transformer hierárquico interpretável
Por que prever as funções das enzimas é importante
Cada célula viva funciona graças a incontáveis pequenas máquinas químicas chamadas enzimas. Cada enzima tem uma “função” específica, e essa função é codificada em um número da Comissão de Enzimas (EC), um código em quatro partes parecido com um endereço postal. Atribuir corretamente números EC é crucial para entender o metabolismo, projetar novos medicamentos, engenharia de microrganismos para produzir combustíveis ou alternativas a plásticos e monitorar como ecossistemas processam substâncias químicas. Mas experimentos para determinar funções enzimáticas são lentos e caros. Este estudo apresenta o HIT-EC, um novo modelo de inteligência artificial que pode prever de modo confiável números EC a partir de sequências de proteínas e ainda explicar por que fez cada previsão.
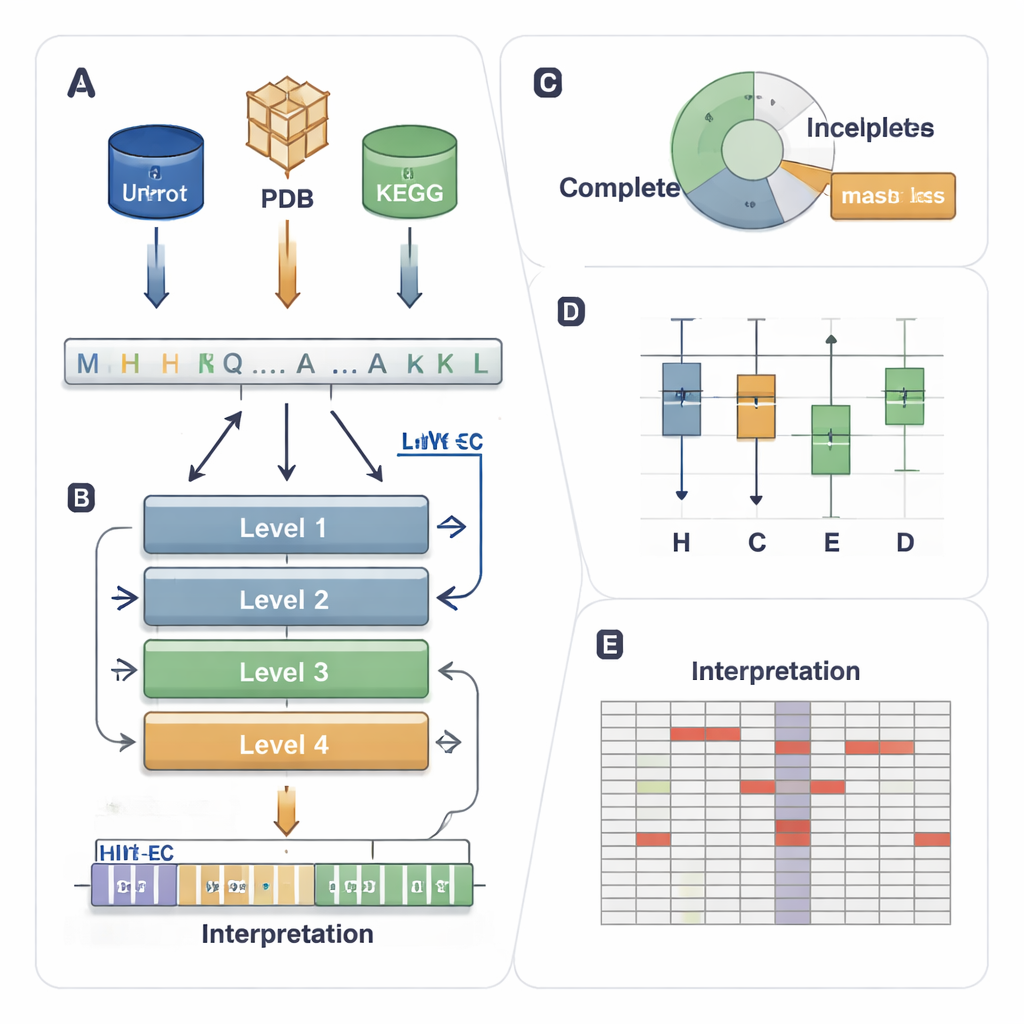
Um sistema de código postal para funções enzimáticas
O sistema EC atribui a cada enzima um código de quatro níveis, por exemplo 1.1.1.37. O primeiro dígito indica uma classe ampla (por exemplo, enzimas que transferem elétrons ou grupos), e os dígitos seguintes descrevem detalhes mais precisos da reação. Essa hierarquia é poderosa, mas gera um problema de previsão exigente: um modelo deve acertar os quatro níveis entre milhares de códigos possíveis, mesmo quando algumas enzimas são raras ou estão apenas parcialmente anotadas em bases de dados (por exemplo, 3.5.-.-, onde os níveis detalhados estão ausentes). Métodos computacionais existentes usam estrutura 3D, similaridade de sequência ou aprendizado profundo, mas tendem a ter dificuldades com enzimas incomuns, ignoram dados parcialmente rotulados e tipicamente funcionam como “caixas-pretas” que oferecem pouca informação sobre o porquê de uma decisão.
Uma IA de quatro andares que segue a escada EC
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) foi projetado para espelhar a hierarquia de quatro níveis do EC. Ele recebe uma sequência bruta de proteína e a processa por quatro camadas transformer, cada uma focada em um nível do EC. Fluxos locais conectam cada nível ao anterior, garantindo que uma decisão de granularidade fina (o quarto dígito) seja consistente com as mais amplas (primeiro e segundo dígitos). Em paralelo, um fluxo global mantém o contexto completo da sequência visível em cada etapa. O modelo também pode ser treinado com sequências que têm rótulos incompletos, usando uma “loss mascarada” que simplesmente ignora níveis EC ausentes em vez de descartar a sequência. Isso permite que o HIT-EC aprenda a partir da grande fração de proteínas em bases curadas que estão apenas parcialmente anotadas.
Superando rivais em precisão e velocidade
Os autores reuniram um grande conjunto de dados cuidadosamente filtrado com cerca de 200.000 enzimas e 1.938 números EC diferentes, provenientes do Swiss-Prot e do Protein Data Bank. Em testes repetidos com hold-out, o HIT-EC superou três métodos de ponta (CLEAN, ECPICK e DeepECtransformer) tanto em F1-score geral quanto em F1 por classe, que medem o equilíbrio entre acertos e alarmes falsos. O modelo foi particularmente forte em códigos EC sub-representados com 25 ou menos exemplos conhecidos, onde métodos anteriores frequentemente falham. O HIT-EC também generalizou bem para enzimas novas adicionadas ao Swiss-Prot após o treinamento e para genomas completos de bactérias diversas, incluindo cepas bem estudadas de Escherichia coli, Bacillus subtilis e Mycobacterium tuberculosis. Apesar de sua sofisticação, o modelo foi altamente eficiente: em uma GPU padrão processou uma proteína em cerca de 38 milissegundos—dezenas de vezes mais rápido que alguns concorrentes que dependem de buscas de similaridade mais lentas ou de ensembles de muitos modelos.
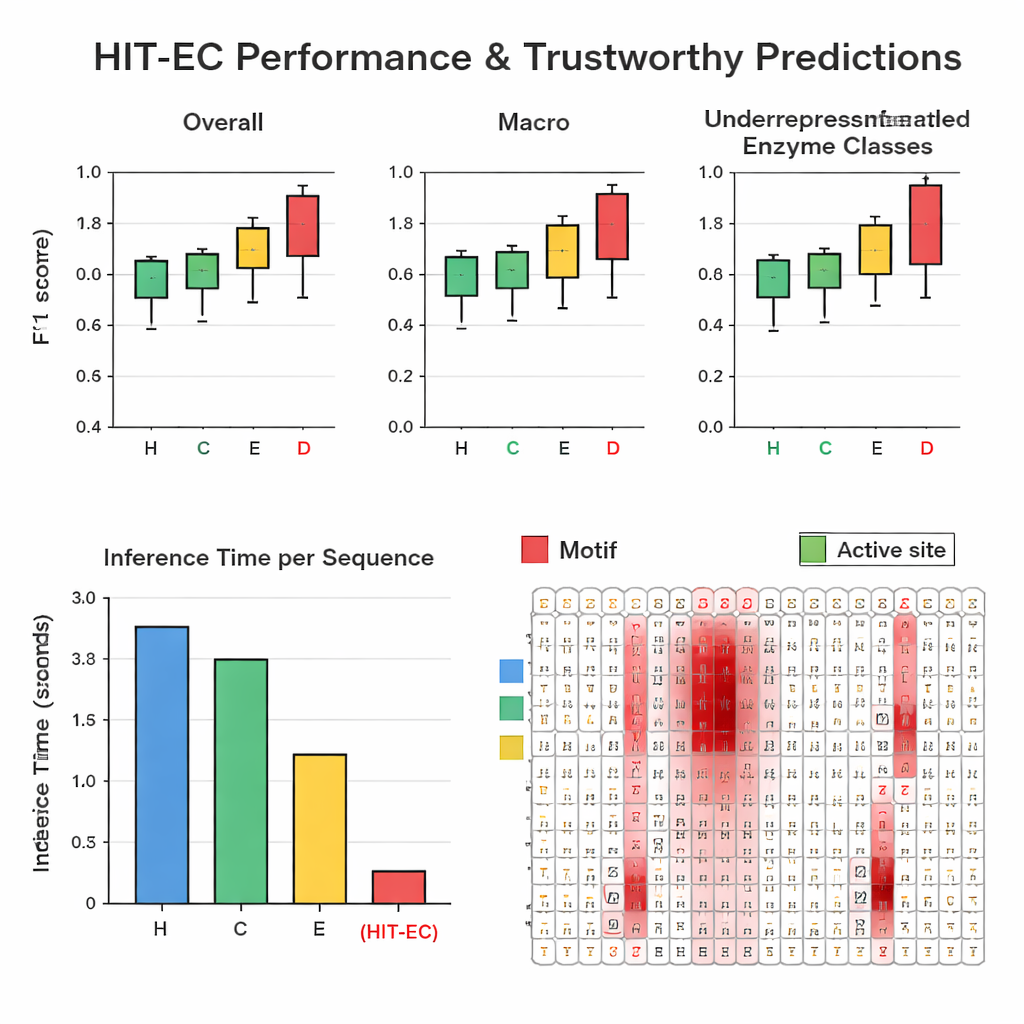
Vendo o que o modelo “olha”
Para tornar suas previsões confiáveis, o HIT-EC foi desenhado para mostrar quais aminoácidos na sequência influenciaram cada decisão em nível EC. Os autores construíram uma via de interpretação que combina pesos de atenção com informação de gradiente para pontuar a importância de cada posição. Eles validaram essas pontuações em famílias enzimáticas bem caracterizadas. Por exemplo, em uma família de citocromo P450 (CYP106A2), o HIT-EC destacou motivos funcionais conhecidos, como regiões de ligação ao oxigênio e ao heme, e identificou um sutil motivo EXXR que um modelo de referência deixou passar. Para representantes clássicos de cada classe EC de topo—como álcool desidrogenase, hexocinase e anidrase carbônica—os escores de relevância do modelo iluminaram motivos característicos de livros-texto e sítios de ligação ao substrato. Essas interpretações fornecem “evidência” bioquímica de que o modelo baseia suas previsões em características significativas, não em correlações acidentais.
Orientando trabalhos sobre enzimas raras e emergentes
A equipe testou ainda o HIT-EC em duas enzimas pouco estudadas importantes para descontaminação: um citocromo P450 envolvido na degradação de poluentes aromáticos e uma hidrolase degradadora de PET de Streptomyces que ajuda a digerir moléculas relacionadas a plásticos. Ambas as enzimas haviam sido caracterizadas experimentalmente, mas não tinham atribuições EC oficiais. O HIT-EC previu corretamente os números EC esperados e destacou padrões de motivos e resíduos catalíticos que correspondem ao que se conhece por estudos estruturais e bioquímicos. No geral, o trabalho mostra que o HIT-EC pode não só atribuir números EC com mais precisão e rapidez do que as ferramentas atuais, especialmente para funções raras, como também esclarecer por que uma enzima é considerada responsável por uma determinada reação química. Essa combinação de desempenho e interpretabilidade faz dele um motor promissor para anotação enzimática confiável e em larga escala em genômica, biotecnologia e pesquisa ambiental.
Citação: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Palavras-chave: previsão de função enzimática, aprendizado profundo em biologia, modelos transformer, anotação de proteínas, enzimas para biorremediação