Clear Sky Science · pt
Uma ativação alternativa do EGFR pela mutação R252C derivada do paciente promove a progressão do câncer
Quando as antenas celulares ficam fora de controle
Por que alguns cânceres continuam crescendo mesmo após ciclos de quimioterapia e imunoterapia de ponta? Este estudo acompanha um paciente com tumores no pulmão e no cérebro e rastreia a doença até uma pequena alteração em uma “antena”-chave da superfície celular chamada EGFR. Ao revelar como essa única mutação reprograma sinais de crescimento, os pesquisadores não apenas explicam o caráter agressivo do câncer do paciente, como também mostram como um medicamento já existente, o afatinibe, pode contê-lo.
Uma mutação rara com grandes consequências
EGFR é um receptor que atravessa a membrana celular e ajuda as células a responderem a sinais de crescimento. Muitos cânceres de pulmão e do cérebro apresentam mutações em EGFR, mas a maioria das alterações conhecidas fica na porção interna da célula, na região que funciona como um interruptor enzimático. Aqui, a equipe descobriu uma alteração incomum na parte externa do EGFR, na região que normalmente se liga a fatores de crescimento. Em um paciente com câncer de pulmão e glioma, encontraram que um aminoácido na posição 252 foi trocado de arginina para cisteína — batizada de EGFR R252C. A mineração de bancos de dados de câncer mostrou essa mutação em uma pequena parcela de pacientes com glioma e quase nunca em tumores de pulmão, sugerindo que é rara, porém real. Usando ferramentas de edição gênica, os autores recriaram exatamente essa mutação em várias linhagens humanas de câncer cerebral e pulmonar para testar sua função.
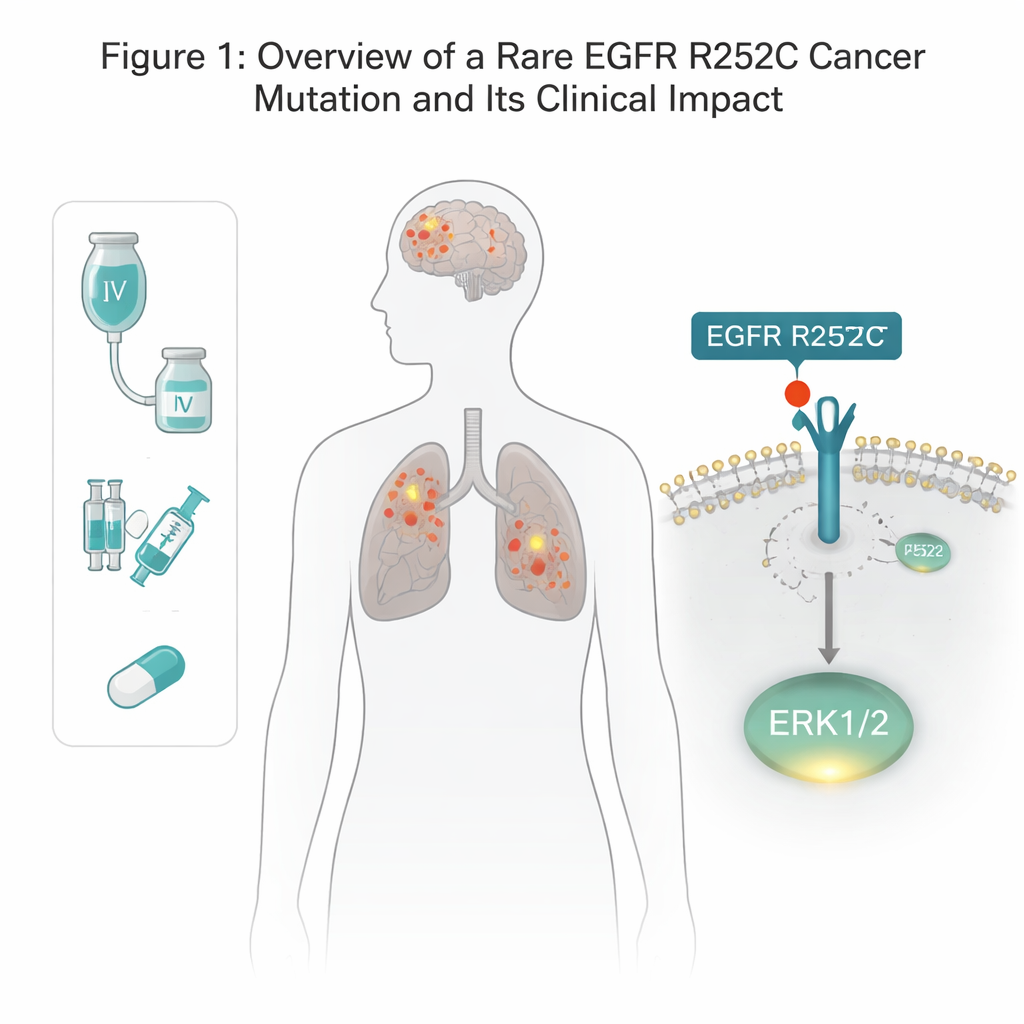
Um novo atalho para os sinais de crescimento
Ordinaryamente, o EGFR precisa parear com outra cópia e então marcar sua própria cauda interna com grupos fosfato antes que possa ativar as vias de crescimento a jusante. Surpreendentemente, a versão R252C não apresentou essa auto-fosforilação habitual. Em vez disso, células que carregavam EGFR R252C ativaram de forma muito mais intensa um controlador específico de crescimento, ERK1/2, enquanto deixavam outras rotas clássicas do EGFR — como AKT e STAT3 — em grande parte inalteradas. Bloquear ERK1/2 com um inibidor dedicado eliminou a vantagem de crescimento extra das células R252C, provando que ERK1/2 é o principal motor por trás do poder tumorigênico dessa mutante.
Trancando o receptor em uma forma sempre ligada
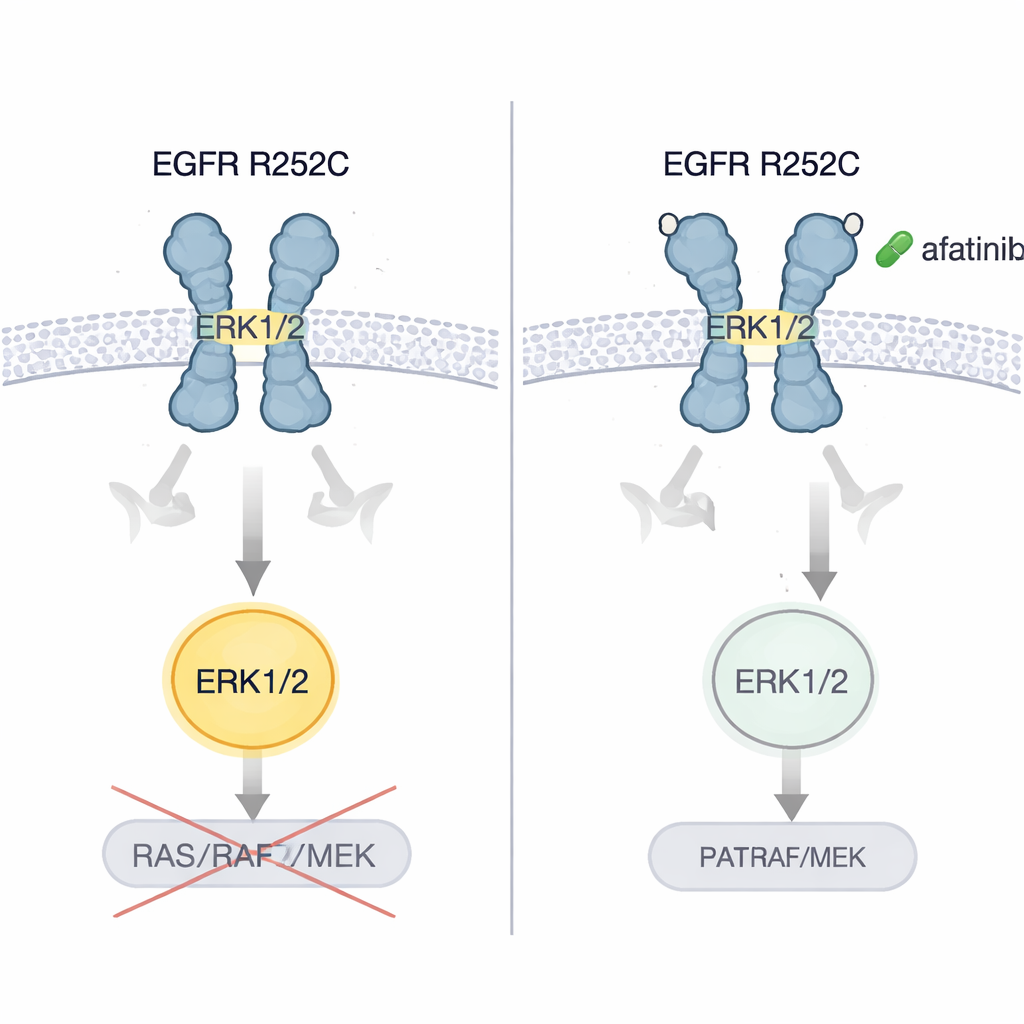
Para entender como uma mudança externa poderia causar essa superativação seletiva, os pesquisadores combinaram ensaios bioquímicos com simulações por computador. A troca R252C introduz uma nova cisteína na porção externa do EGFR. Duas dessas mutantes podem formar uma ligação dissulfeto — uma espécie de grampo molecular — entre seus resíduos C252, prendendo-se uma à outra em um par estável. A modelagem estrutural mostrou que essa ligação força a parte externa do receptor a assumir um alinhamento escalonado em forma de “V” que imita de perto a conformação ativa ligada ao ligante, mesmo na ausência de fator de crescimento. Esse alinhamento se propaga através dos segmentos transmembrana e da região logo dentro da membrana, torcendo os domínios enzimáticos internos em uma disposição incomum: os sítios ativos ficam voltados para dentro da célula, mas mantidos afastados demais para fosforilarem eficientemente entre si. Em vez disso, essa conformação cria uma superfície de ancoragem forte para ERK1/2, permitindo que o EGFR R252C fosforile diretamente ERK1/2 e contorne o habitual circuito RAS–RAF–MEK.
De modelos em camundongos a um único paciente
Os autores demonstraram que células de câncer cerebral e pulmonar portadoras de EGFR R252C cresceram mais rapidamente em placas e formaram tumores maiores e mais agressivos quando implantadas em camundongos, em comparação com células que carregavam EGFR normal. Em seguida testaram várias gerações de pílulas que bloqueiam o EGFR. Apenas o afatinibe, um inibidor de segunda geração, suprimiu de forma consistente a ativação de ERK1/2 e reduziu fortemente o crescimento das células tumorais. Em modelos murinos de tumores cerebrais e pulmonares conduzidos por R252C, o afatinibe retardou a expansão tumoral e estendeu a sobrevida. De forma crítica, quando o paciente original — cuja doença havia piorado apesar da quimioterapia, de um fármaco direcionado a vasos sanguíneos e da imunoterapia — foi tratado com afatinibe, as imagens do pulmão e do cérebro mostraram redução marcante da carga tumoral e o paciente teve vários anos sem progressão.
O que isso significa para os pacientes
Este trabalho revela uma maneira até então não reconhecida pela qual uma mutação oncogênica do EGFR pode operar: ao grampear dois receptores juntos fora da célula, torcê‑los em uma pose ativa que liga diretamente o ERK1/2 em vez de seguir a cadeia de sinalização dos livros-texto. Para não especialistas, a conclusão principal é que nem todas as mutações no mesmo gene se comportam da mesma forma, e algumas alterações raras podem responder melhor a fármacos específicos já existentes. O EGFR R252C parece gerar cânceres particularmente vulneráveis ao afatinibe. Embora essa conclusão atualmente se apoie em um caso clínico detalhado mais um extenso trabalho de laboratório, ela aponta para testes mais personalizados de mutações do domínio externo do EGFR e sugere que terapias-alvo cuidadosamente escolhidas podem oferecer nova esperança a pacientes selecionados com tumores cerebrais e pulmonares de difícil tratamento.
Citação: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Palavras-chave: mutação EGFR, glioma, câncer de pulmão, sinalização ERK, afatinibe