Clear Sky Science · pt
Estimativa da estabilidade do dobramento de proteínas com consideração explícita dos estados desnaturados
Por que a estabilidade das proteínas importa
Cada proteína no seu corpo é uma pequena máquina molecular que precisa se dobrar em uma forma tridimensional precisa para funcionar corretamente. Se esse enovelamento for frágil demais, a proteína pode falhar, agrupar‑se ou deixar de ser produzida — problemas associados a doenças e a falhas na fabricação de medicamentos e enzimas à base de proteínas. Medir quão estável uma proteína é no laboratório é um processo lento e complexo, por isso os cientistas buscam métodos computacionais que possam indicar, apenas a partir da sequência, com que facilidade uma proteína se desnaturará.
Um novo olhar sobre estados dobrados e desnaturados
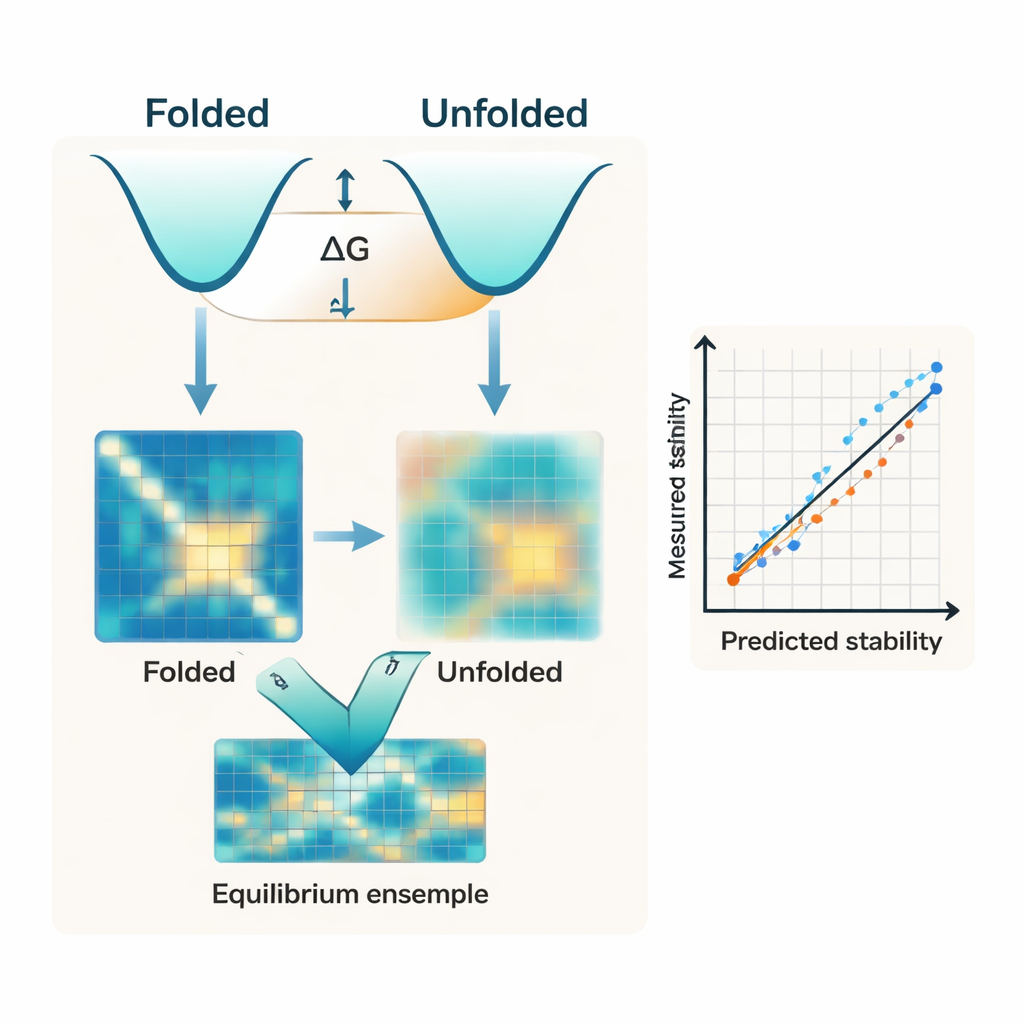
A maioria dos algoritmos modernos foca quase inteiramente na conformação dobrada da proteína. Eles frequentemente partem de uma estrutura prevista por IA, como as do AlphaFold, e tratam essa única estrutura como o principal determinante da estabilidade. Mas a estabilidade é, na verdade, a lacuna de energia entre dois conjuntos amplos: o estado compacto dobrado e as muitas formas flexíveis que compõem o estado desnaturado. Os autores argumentam que ignorar o lado desnaturado desse balanço é uma razão central pela qual ferramentas existentes têm dificuldade em corresponder às medições experimentais da energia livre de dobramento, conhecida como ΔG.
Um novo modelo que aprende ambos os estados
Os pesquisadores apresentam o IFUM, um sistema de aprendizado profundo projetado para estimar ΔG enquanto também aprende como é o equilíbrio entre os estados dobrado e desnaturado para cada proteína. Em vez de tratar o estado desnaturado como um pano de fundo vago, o IFUM usa ideias da física de polímeros para representá‑lo como uma “espiral aleatória” e codifica ambos os estados — dobrado e desnaturado — como mapas de distâncias entre pares de aminoácidos. O modelo incorpora informações de potentes redes pré‑treinadas de sequência e estrutura e, em seguida, prevê em conjunto a estabilidade total e um mapa de probabilidades que descreve quanto da população proteica está dobrada versus desnaturada em cada par de resíduos. O treinamento em um conjunto de dados muito grande de proteínas pequenas caracterizadas experimentalmente e de proteínas desordenadas conhecidas ajuda o IFUM a reconhecer tanto sequências bem estruturadas quanto sequências flexíveis.
Números melhores e maior cobertura de mutações
Quando testado em um conjunto de dados cuidadosamente controlado de proteínas pequenas, o IFUM prevê valores experimentais de ΔG com menor erro e maior correlação do que métodos baseados em IA anteriores que dependem apenas da estrutura dobrada ou de modelos de linguagem treinados em sequências. De forma crucial, o modelo também lida com uma ampla variedade de alterações de sequência. Ele captura com precisão os efeitos de mutações pontuais simples e duplas, assim como inserções e deleções que alteram o comprimento da proteína — situações em que muitas ferramentas existentes ou falham completamente ou não foram projetadas para operar. Uma comparação interna mostra que remover o objetivo relativo ao estado desnaturado piora significativamente o desempenho, reforçando que modelar explicitamente o conjunto desnaturado não é apenas uma elegância conceitual, mas central para a precisão das previsões.
Do banco de design aos testes no mundo real
Para verificar se o IFUM pode orientar engenharia protéica real, os autores o aplicam a três problemas de design desafiadores: estabilizar proteínas interferon‑lambda, remodelar a proteína de sinalização imune IL‑10 e melhorar uma enzima modificadora de açúcares chamada UGT76G1. Em todos os três casos, as estabilidade previstas pelo IFUM acompanham bem as temperaturas de fusão medidas, que indicam quanta temperatura uma proteína suporta antes de se desnaturar. O modelo também ajuda a triagem de centenas de proteínas completamente novas, projetadas por computador, para selecionar aquelas com maior probabilidade de se dobrar e permanecer solúveis em células, superando scores de confiança amplamente usados por redes de predição de estrutura. Esses resultados sugerem que o IFUM pode ser usado como um “filtro de estabilidade” prático junto com verificações baseadas em estrutura em fluxos de trabalho modernos de design de proteínas.
Limites e direções futuras
Como qualquer modelo, o IFUM tem limites. Ele é treinado principalmente em proteínas curtas, de cadeia única e solúveis, e seus números absolutos de estabilidade tornam‑se menos confiáveis para proteínas muito maiores ou para aquelas com longas alças flexíveis ou regiões transmembranares extensas. Sua descrição do estado desnaturado ainda é um modelo estatístico simplificado, em vez de uma representação totalmente realista de todas as formas possíveis. Ainda assim, a abordagem demonstra que ensinar uma IA a considerar tanto os conjuntos dobrados quanto os desnaturados produz estimativas de estabilidade mais confiáveis. Para não especialistas, a conclusão principal é que o IFUM nos aproxima de poder perguntar a um computador, com confiança quantitativa, “Este design de proteína realmente vai se manter?”, potencialmente acelerando o desenvolvimento de medicamentos biológicos mais seguros e de enzimas industriais mais robustas.
Citação: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Palavras-chave: estabilidade de proteínas, dobramento de proteínas, aprendizado profundo, design de proteínas, mutações