Clear Sky Science · pt
Benchmarking abrangente de métodos de escores poligênicos mona e multi ancestral com a plataforma PGS-hub
Por que sua pontuação de risco de DNA importa
Os médicos estão ficando melhores em ler nosso DNA para estimar quem tem maior probabilidade de desenvolver doenças comuns, como doenças cardíacas, diabetes ou esquizofrenia. Essas estimativas, chamadas escores poligênicos, combinam os efeitos pequenos de muitas variantes genéticas em um único número. Mas hoje existem várias maneiras concorrentes de calcular esses escores, e elas não funcionam igualmente bem para pessoas de diferentes origens ancestrais. Este estudo teve como objetivo comparar métodos líderes frente a frente e construir um serviço online, o PGS-hub, que permite aos pesquisadores calcular esses escores de forma consistente e simples.
Um ponto único para calculadoras de risco genético
Os autores criaram o PGS-hub, uma plataforma web que oculta grande parte da complexidade técnica por trás dos escores poligênicos. Usuários carregam resultados de estudos genéticos que resumem como milhões de marcadores de DNA se relacionam com uma doença ou traço. Em seguida, escolhem o fundo ancestral da população de interesse — por exemplo, europeu ou africano — e selecionam a partir de um menu de métodos de pontuação populares. Nos bastidores, o PGS-hub converte a entrada nos formatos apropriados, conecta painéis de referência pré-construídos que descrevem como marcadores próximos do DNA estão correlacionados e executa um grande número de tarefas em um sistema de computação de alto desempenho. O resultado é um arquivo compacto de pesos que pode ser aplicado a genomas individuais para gerar uma pontuação para cada pessoa. 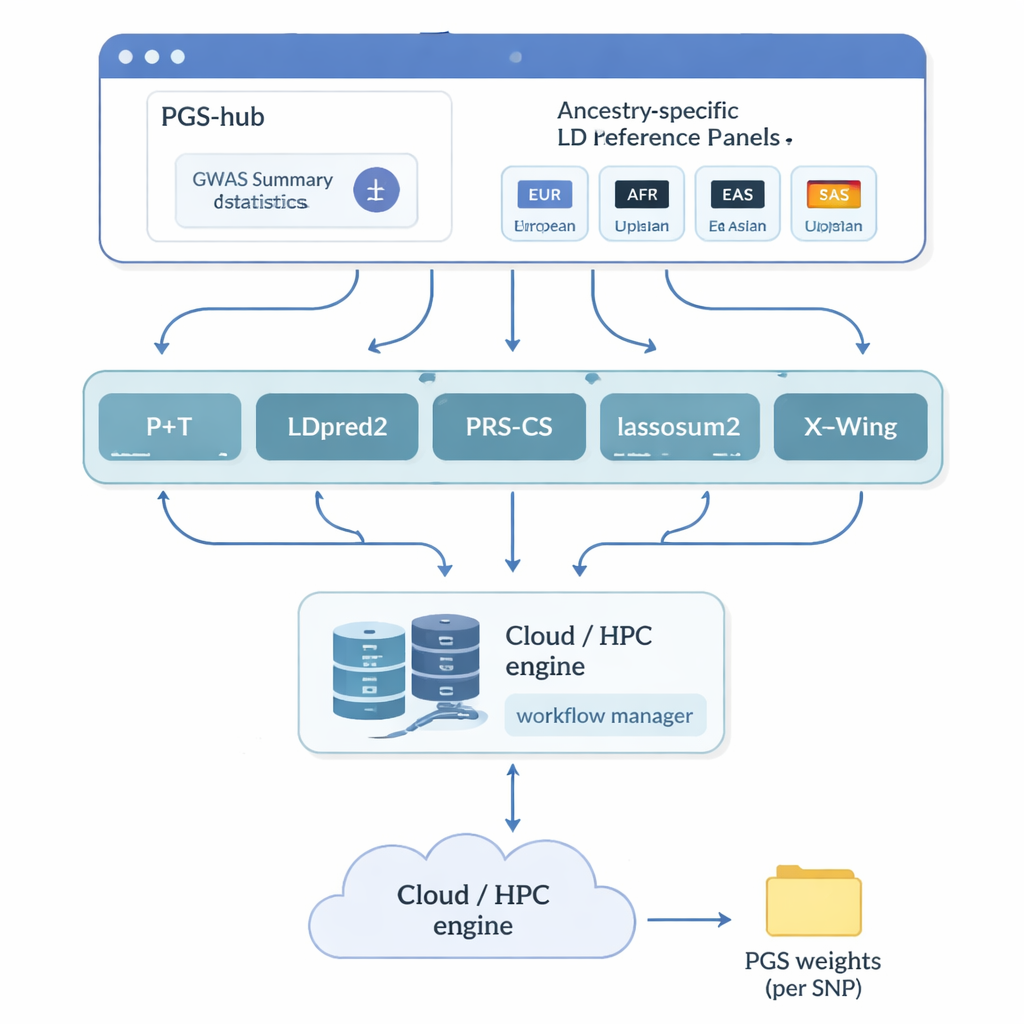
Testando 13 métodos de pontuação
Para verificar quais abordagens funcionam melhor, a equipe comparou 13 métodos de ponta ao longo de 36 doenças e traços em quase 380.000 pessoas de ascendência europeia e pouco mais de 8.000 pessoas de ascendência africana do UK Biobank. Avaliaram não apenas quão bem cada escore previa quem tinha uma doença ou um valor de traço mais alto, mas também quanto tempo de computação e memória cada método consumia. Em europeus, um método chamado LDpred2 geralmente entregou os escores mais precisos, frequentemente superando os demais por margem clara. Algumas alternativas — lassosum2, PRS-CS e SDPR — apresentaram desempenho quase tão bom para muitos traços, enquanto alguns métodos mais antigos ficaram atrás. Para traços como altura ou doença de Crohn, os melhores escores explicaram uma parcela considerável do risco genético; para outros, como função renal, todos os métodos tiveram dificuldade, refletindo sinais genéticos subjacentes mais fracos.
Percepções para populações diversas e métodos combinados
Uma preocupação importante na predição genética é que métodos treinados majoritariamente em europeus podem não se transferir bem para pessoas com ancestrais diferentes. Quando os autores repetiram seus benchmarks usando estudos genéticos de ascendência africana, todos os métodos performaram pior, evidenciando a falta de estudos de grande porte nessas populações. Ainda assim, LDpred2 e SDPR tendiam a estar entre as melhores opções. A equipe também examinou abordagens “multi-ancestrais” que combinam explicitamente informações entre populações. Aqui, uma estratégia relativamente simples — combinar linearmente os melhores escores específicos por ancestralidade do LDpred2 em um único escore LDpred2-multi — superou modelos multi-ancestrais mais elaborados, como PRS-CSx e X-Wing, tanto para grupos europeus quanto africanos. Além disso, os autores mostraram que construir um ensemble, que mistura os escores mais fortes de vários métodos, aumentou ainda mais a predição para todos os traços, especialmente para doenças altamente hereditárias como esquizofrenia e doença arterial coronariana. 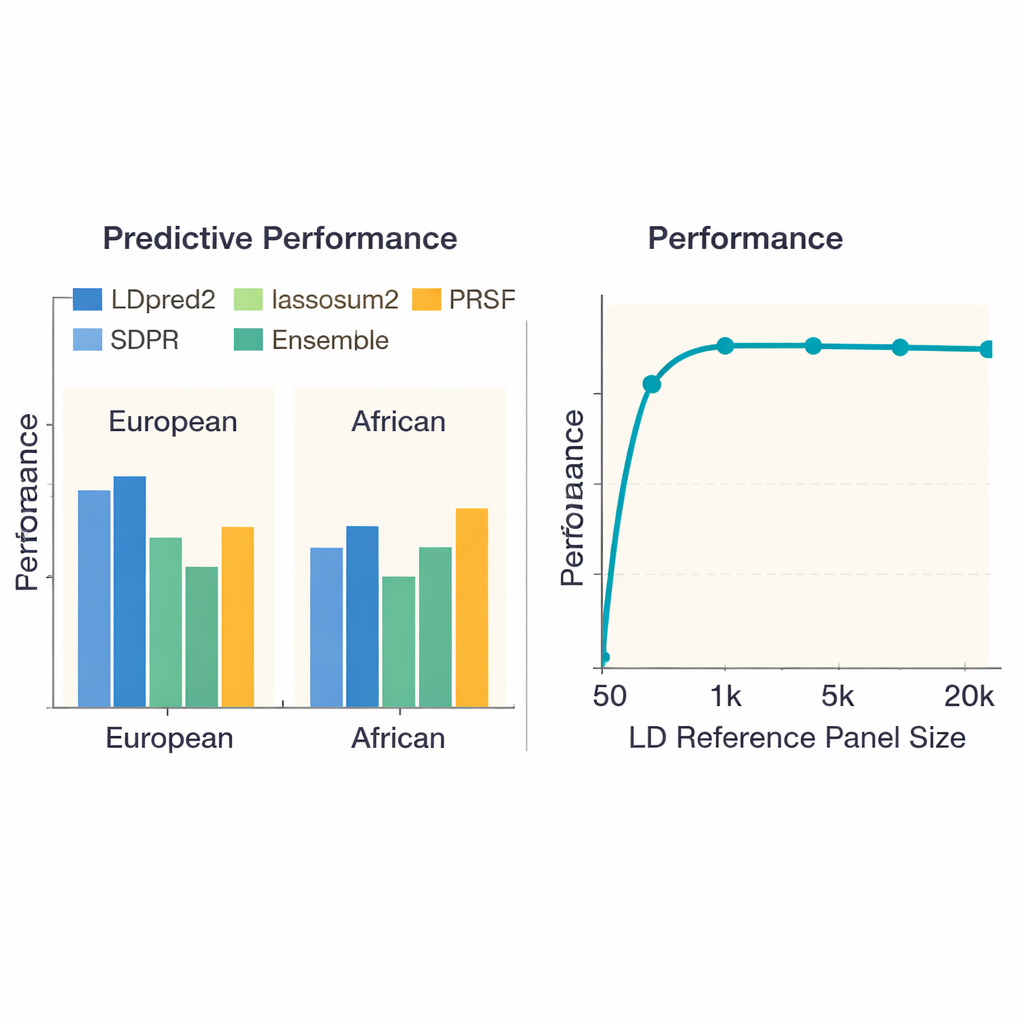
Como escolhas de dados e limites de computação moldam os escores
O estudo investigou como o tamanho do painel de referência — o conjunto de pessoas usado para aprender como marcadores próximos do DNA co-variam — afeta o desempenho. Quando esse painel era muito pequeno (menos de 1.000 indivíduos), os escores eram claramente menos precisos. À medida que o painel crescia para cerca de 5.000 pessoas, o desempenho melhorou fortemente e então se estabilizou, sugerindo que painéis cada vez maiores trazem retornos decrescentes. Surpreendentemente, simplesmente adicionar mais marcadores de DNA nem sempre ajudou: usar cerca de 6,6 milhões de variantes às vezes tornou as predições piores do que usar um conjunto cuidadosamente escolhido de aproximadamente 1,1 milhão, provavelmente porque marcadores extras adicionavam mais ruído do que sinal útil. Os autores também documentaram grandes diferenças no custo computacional. Métodos simples, como o tradicional pruning-and-thresholding, terminavam em menos de uma hora por traço, enquanto algumas abordagens bayesianas requeriam centenas de horas de CPU — informação que importa para projetos grandes ou grupos com recursos limitados.
O que isso significa para a predição futura baseada em DNA
Para não especialistas, a mensagem central é que nem todos os escores de risco de DNA são iguais, e os detalhes de como são construídos influenciam fortemente quem se beneficia deles. Este trabalho oferece orientação prática: métodos como LDpred2 e ensembles bem concebidos tendem a fornecer as predições mais confiáveis em grandes conjuntos de dados europeus, e combinações multi-ancestrais podem superar modelos transpopulacionais mais complexos. Ao mesmo tempo, a queda na precisão para indivíduos de ascendência africana ressalta a necessidade urgente de estudos genéticos maiores e mais diversos. Ao agrupar muitos métodos em uma única plataforma online padronizada, o PGS-hub reduz a barreira para que pesquisadores em todo o mundo gerem e comparem escores poligênicos — um passo importante para usar esses escores de forma justa e eficaz na medicina.
Citação: Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Palavras-chave: escores poligênicos, predição de risco genético, plataforma PGS-hub, genômica multi-ancestral, UK Biobank