Clear Sky Science · pt
Modelo de linguagem genômica reduz artefatos de quimera no sequenciamento direto de RNA por nanopore
Por que limpar leituras de RNA é importante
Nossas células estão constantemente lendo instruções genéticas escritas em RNA, e tecnologias de sequenciamento recentes permitem aos cientistas observar esse processo com um nível de detalhe sem precedentes. Uma das ferramentas mais poderosas, o sequenciamento direto de RNA por nanopore, pode ler moléculas de RNA inteiras de uma só vez — mas também introduz falhas que podem fazer parecer que genes estão quebrados e reconstituídos de maneiras que não ocorrem na realidade. Este estudo apresenta o DeepChopper, uma ferramenta de software que funciona como um modelo de linguagem para genomas, corrigindo esses erros para que os pesquisadores possam confiar no que veem nos dados de RNA.
Quando o sequenciador inventa fusões gênicas falsas
As máquinas modernas de nanopore puxam fios individuais de RNA por poros minúsculos e leem sua sequência diretamente. Isso traz grandes vantagens em relação a métodos antigos, como preservar modificações químicas e capturar transcritos em comprimento total em uma única leitura. Mas o processo também depende de pequenos fragmentos auxiliares chamados adaptadores, que são colados às moléculas de RNA durante a preparação da biblioteca. Às vezes, duas ou mais moléculas de RNA são acidentalmente unidas por esses adaptadores, criando o que parecem ser quimeras — moléculas híbridas que aparentam fundir genes diferentes. Ferramentas de análise padrão podem interpretar mal esses resquícios técnicos como eventos biológicos reais, como fusões gênicas relacionadas ao câncer ou padrões de emenda incomuns, levando a resultados enganosos.
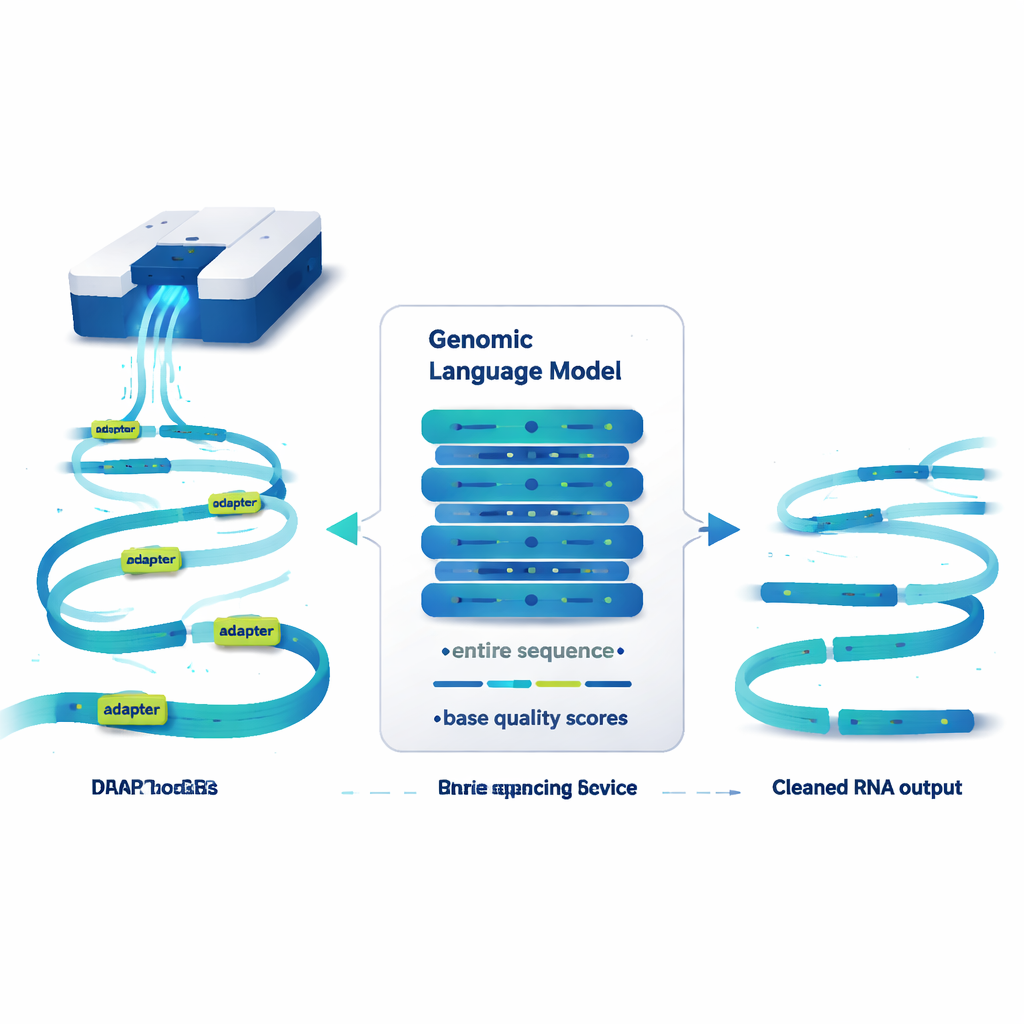
Um modelo de linguagem que lê genomas, não frases
O DeepChopper trata sequências genéticas de forma semelhante a um texto e aplica ideias de grandes modelos de linguagem a elas. Em vez de trabalhar com palavras, ele lê sequências de RNA uma letra por vez, juntamente com uma pontuação de qualidade para cada letra que indica quão confiável é a leitura. Construído sobre uma arquitetura compacta chamada HyenaDNA, ele pode escanear até 32.000 bases de uma só vez — comprimento suficiente para cobrir praticamente qualquer molécula de RNA humana. Para cada posição, o DeepChopper estima se essa base faz parte de uma sequência de RNA genuína ou de um adaptador. Uma etapa de refinamento então suaviza essas previsões para que os adaptadores sejam marcados como blocos contínuos em vez de pontos espalhados.
Cortando as junções ruins sem desperdiçar dados
Uma vez que o DeepChopper identifica adaptadores dentro de uma leitura, ele faz algo crucial: em vez de descartar a leitura inteira, ele “corta” nesses pontos de adaptador e mantém os fragmentos reais. Dessa forma, uma fusão artificial de dois RNAs pode ser separada de volta em suas partes originais. Em testes com milhões de leituras de nanopore de várias linhagens celulares humanas de câncer e de células-tronco, o DeepChopper superou amplamente as ferramentas de remoção de adaptadores existentes, que não foram projetadas para este contexto de RNA direto. Ele reconheceu adaptadores com mais de 99% de precisão e cobertura em benchmarks sintéticos, e escalou eficientemente para conjuntos de dados com mais de 20 milhões de leituras usando processadores gráficos.
Separando fusões gênicas reais de miragens do sequenciamento
Os autores então investigaram se o DeepChopper poderia distinguir eventos biológicos genuínos de artefatos em dados reais de câncer. Ao comparar leituras de RNA direto com conjuntos de dados correspondentes produzidos por métodos independentes (como sequenciamento direto de cDNA em plataformas Oxford Nanopore e PacBio), eles puderam rotular quais quimeras aparentes eram suportadas por outras tecnologias e quais não eram. O DeepChopper reduziu alinhamentos quiméricos não suportados em até 62–91%, ao mesmo tempo que aumentou significativamente a fração confirmada por outros métodos. Também reduziu o número de chamadas suspeitas de fusão gênica em quase 90%, especialmente aquelas envolvendo genes ribossomais que se mostraram artefatos frequentes. Ao mesmo tempo, eventos de fusão verdadeiros respaldados por sequenciamento de RNA de leitura curta foram preservados.
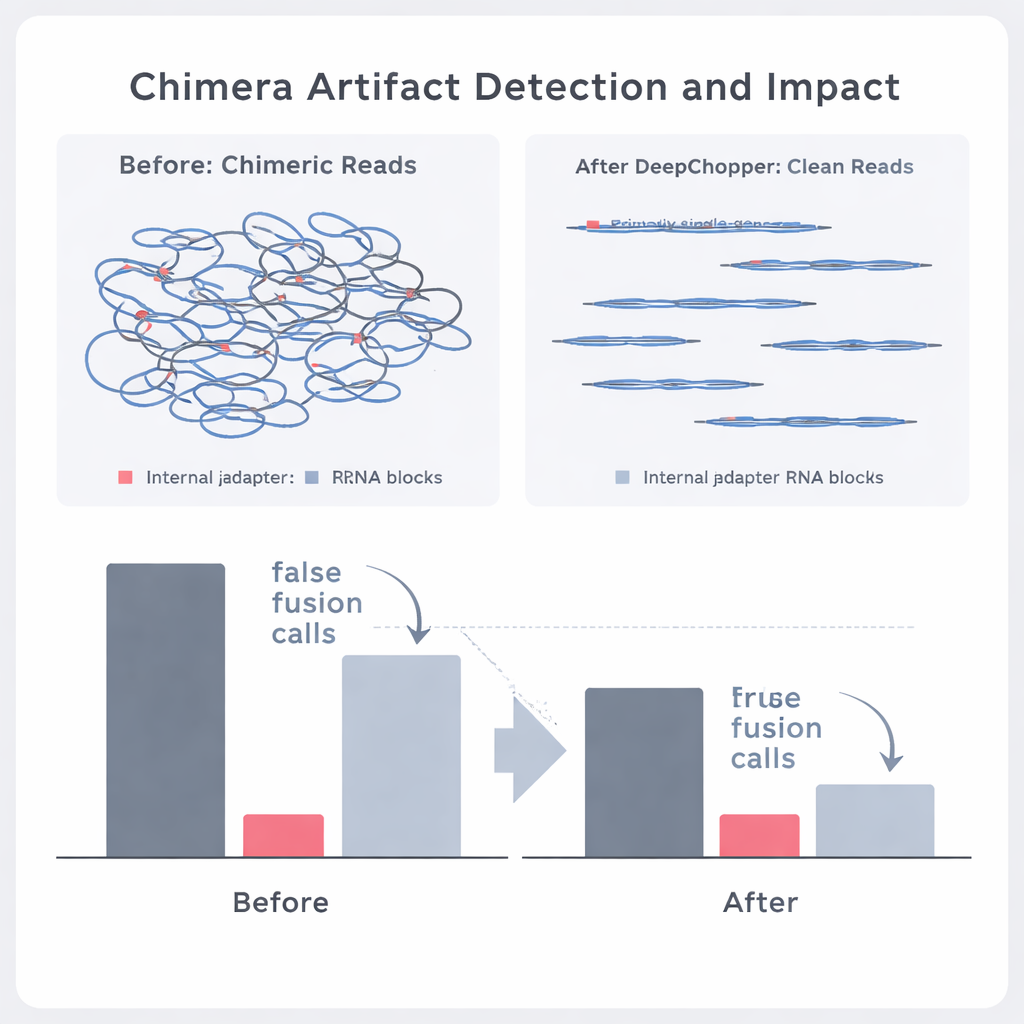
Química melhor ajuda — mas artefatos persistem
A Oxford Nanopore lançou recentemente um kit de sequenciamento atualizado (RNA004) projetado em parte para reduzir artefatos técnicos. O DeepChopper foi aplicado “pronto para uso” a dados dessa nova química e ainda assim encontrou que uma fração pequena, mas importante, das leituras continha adaptadores internos e junções quiméricas. Mesmo sem treinamento extra, o modelo cortou quimeras artefatuais em cerca de um quinto; após ajuste fino com os novos dados, seu desempenho melhorou ligeiramente, mantendo os sinais genuínos intactos. Em todas as químicas e tipos celulares, corrigir esses artefatos permitiu que ferramentas subsequentes detectassem muito mais transcritos em comprimento total e variantes alternativas, oferecendo uma visão mais clara do panorama do RNA celular.
O que isso significa para estudos futuros de RNA
Para não especialistas, a mensagem principal é que nem toda conexão surpreendente de RNA reportada por um sequenciador representa biologia real — algumas são erros introduzidos pela própria tecnologia. O DeepChopper age como um revisor altamente treinado das leituras de RNA por nanopore, identificando as sequências de adaptador que unem moléculas não relacionadas e cortando-as com precisão de base única. O resultado são mapas mais limpos e confiáveis de quais moléculas de RNA existem em uma célula e de como elas são montadas. À medida que laboratórios dependem cada vez mais do sequenciamento de RNA de leitura longa para estudar câncer, distúrbios cerebrais e outras doenças complexas, ferramentas como o DeepChopper serão essenciais para transformar leituras brutas e ruidosas em percepções biológicas confiáveis.
Citação: Li, Y., Wang, TY., Guo, Q. et al. Genomic language model mitigates chimera artifacts in nanopore direct RNA sequencing. Nat Commun 17, 1864 (2026). https://doi.org/10.1038/s41467-026-68571-5
Palavras-chave: sequenciamento de RNA por nanopore, leituras quiméricas, artefatos de fusão gênica, modelo de linguagem genômica, DeepChopper