Clear Sky Science · pt
TET1 como regulador mestre controlando a vigilância da ferroptose dependente e independente de GPX4 na leucemia mieloide aguda
Por que esta pesquisa importa para o tratamento do câncer
Muitos novos medicamentos contra o câncer procuram induzir nas células malignas um modo de autodestruição chamado ferroptose, um tipo de morte celular movida por ferro e dano lipídico. No entanto, alguns tumores resistem persistentemente a essa abordagem. Este estudo revela como uma proteína que modifica o DNA chamada TET1 ajuda células leucêmicas a escapar da ferroptose por meio de dois sistemas bioquímicos de defesa distintos — e mostra que bloquear essas defesas pode tornar vulneráveis até mesmo cânceres resistentes.
Uma mistura letal de ferro e lipídios danificados
A ferroptose ocorre quando o ferro alimenta a oxidação descontrolada de lipídios nas membranas celulares, levando por fim à ruptura das células. Na leucemia mieloide aguda (LMA), como em muitos tipos de câncer, as células empregam sistemas de vigilância poderosos para manter esse processo sob controle. Um guardião chave é a enzima GPX4, que utiliza uma pequena molécula chamada glutationa para neutralizar peróxidos lipídicos nocivos. Outros sistemas de reserva geram moléculas antioxidantes que podem aprisionar radicais perigosos mesmo quando a GPX4 está comprometida. Entender quais interruptores mestres coordenam essas defesas é crucial para projetar terapias que provoquem ferroptose de forma confiável nas células cancerosas, poupando os tecidos saudáveis.
TET1 surge como um centro de controle central
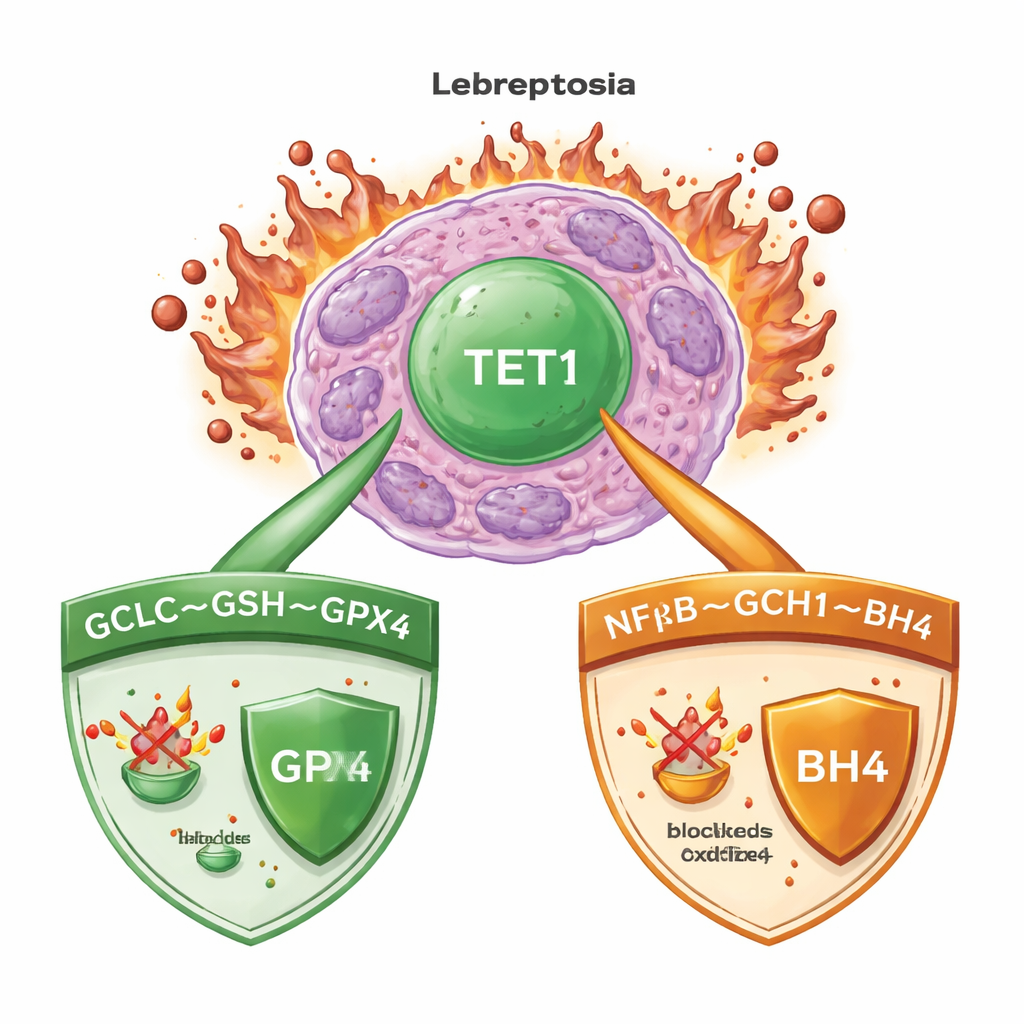
Os pesquisadores compararam dezenas de amostras de células cancerosas, incluindo várias linhagens de LMA e células derivadas de pacientes, e notaram um padrão claro: células que resistiam à ferroptose apresentavam níveis mais altos de TET1, uma enzima que altera marcas químicas do DNA e influencia a atividade gênica. Quando reduziram os níveis de TET1 usando ferramentas genéticas ou inibiram sua atividade com uma pequena molécula, as células cancerosas tornaram-se marcadamente mais sensíveis a fármacos que induzem ferroptose. Isso se manteve tanto em culturas quanto em modelos murinos de LMA. Por outro lado, aumentar a expressão de TET1 protegeu as células da morte ferroptótica e limitou o acúmulo de espécies reativas de oxigênio, os subprodutos quimicamente agressivos que promovem o dano da membrana.
Fortalecendo o principal escudo antioxidante
Aprofundando, a equipe mapeou onde o TET1 atua no genoma e descobriu que ele ativa diretamente um gene chamado GCLC. GCLC codifica uma enzima crítica que inicia a produção de glutationa, o combustível para a GPX4. Ao aumentar uma marca específica do DNA (5-hidroximetilcitosina) no promotor de GCLC, o TET1 eleva a síntese de glutationa. Em condições nutritivas normais, isso amplia a reserva antioxidante principal da célula. Sob privação de cistina, o mesmo complexo enzimático também gera peptídeos incomuns γ-glutamil que ajudam a eliminar o excesso de glutamato, outra maneira de atenuar a ferroptose. Tanto em células cultivadas quanto em camundongos, a perda de TET1 ou a inibição farmacológica da síntese de glutationa reduziram drasticamente os níveis de glutationa e desses peptídeos protetores, tornando as células leucêmicas muito mais vulneráveis aos indutores de ferroptose.
Uma segunda rota de fuga independente de GPX4
Surpreendentemente, o papel protetor do TET1 não se limitou ao eixo glutationa–GPX4. Mesmo quando a própria GPX4 foi removida das células leucêmicas, o excesso de TET1 ainda podia prevenir a morte ferroptótica, sugerindo uma segunda linha de defesa. Os autores rastrearam isso até a ativação pelo TET1 da via de sinalização NFκB, em particular um componente chamado NFKB2. Isso, por sua vez, aumenta a expressão de GCH1, uma enzima que produz a molécula antioxidante BH4. A BH4 pode proteger os lipídios da membrana da oxidação sem depender da GPX4. Quando GCH1 foi silenciado geneticamente ou bloqueado quimicamente, a capacidade do TET1 de proteger as células da ferroptose foi parcialmente perdida. Juntos, esses achados definem uma via TET1–NFKB2–GCH1 que forma um sistema de vigilância da ferroptose independente de GPX4.
Transformando uma fraqueza em oportunidade terapêutica
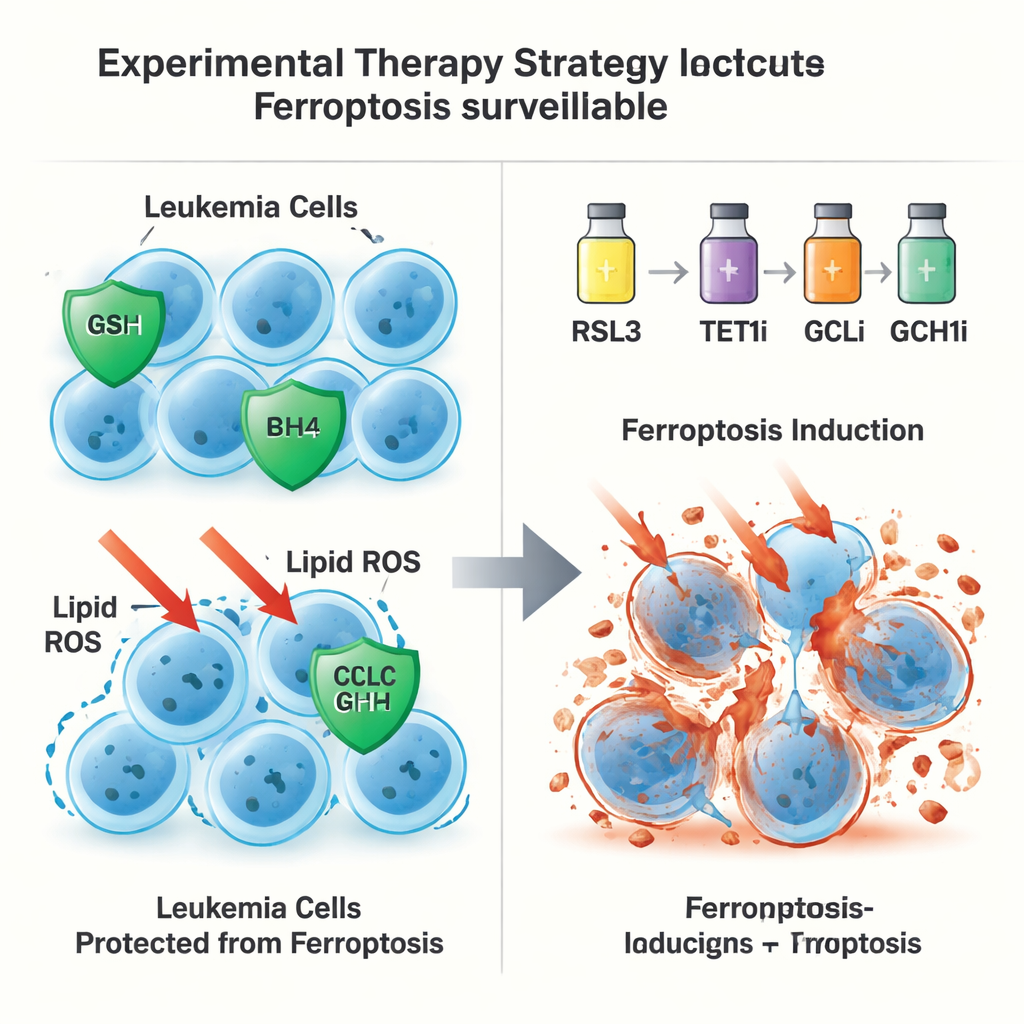
Munidos desse mapa de dupla via, os pesquisadores testaram se, simultaneamente, empurrar a ferroptose e enfraquecer as defesas controladas por TET1 poderia oferecer uma vantagem terapêutica. Em modelos murinos de LMA e enxertos de leucemia derivados de pacientes em camundongos, doses baixas de um fármaco indutor de ferroptose combinadas com inibidores de TET1, da síntese de GSH (via GCLC) ou de GCH1 reduziram dramaticamente a carga leucêmica, prolongaram a sobrevida e esgotaram populações de células iniciadoras de leucemia. Importante, o indutor de ferroptose foi usado em uma fração das doses relatadas em estudos anteriores, diminuindo preocupações sobre toxicidade para células-tronco sanguíneas normais.
O que isso significa para futuras terapias contra o câncer
Para não especialistas, a mensagem principal é que as células leucêmicas sobrevivem executando dois sistemas de “escudo” antioxidante sobrepostos, ambos coordenados pelo TET1: um centrado na glutationa e na GPX4, e outro baseado em GCH1 e BH4. Este trabalho mostra que, ao ativar modestamente a ferroptose enquanto bloqueia o TET1 e seus parceiros a jusante, médicos poderão um dia superar resistência e empurrar seletivamente as células cancerosas além do limite, sem sobrecarregar os tecidos saudáveis. Embora essas estratégias ainda não estejam prontas para a clínica, o estudo identifica o TET1 como um nó de controle poderoso e um alvo promissor para terapias combinadas na LMA e potencialmente em outros cânceres difíceis de tratar.
Citação: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Palavras-chave: ferroptose, leucemia mieloide aguda, TET1, glutationa, epigenética do câncer