Cânceres de pulmão resistentes a inibidores de EGFR exibem sensibilidade colateral a uma ponte molecular covalente e oligomerizante de KEAP1 independente de cisteína
Por que o câncer de pulmão resistente a drogas importa
Fármacos direcionados transformaram o tratamento de certos cânceres de pulmão ao atacar um sinal de crescimento defeituoso chamado EGFR. No entanto, para a maioria dos pacientes, esses medicamentos deixam de funcionar dentro de alguns anos à medida que o câncer desenvolve resistência. Este estudo revela uma reviravolta surpreendente: quando os tumores se tornam resistentes a inibidores de EGFR, eles desenvolvem um novo calcanhar de Aquiles que pode ser atacado com um tipo diferente de composto. Entender essa fraqueza oculta pode inspirar estratégias terapêuticas futuras que encurralem a evolução do câncer em vez de sempre persegui-la.
Uma fraqueza oculta revelada
Os pesquisadores concentraram-se em cânceres de pulmão de não pequenas células impulsionados por EGFR mutante, uma forma comum da doença. Em laboratório, compararam células cancerígenas sensíveis a drogas com células intimamente relacionadas que haviam evoluído resistência a medicamentos bloqueadores de EGFR como gefitinibe e osimertinibe. Em seguida, testaram uma biblioteca de cerca de 2.100 pequenas moléculas para ver quais matavam as células resistentes de maneira mais eficaz do que as células sensíveis originais. Entre muitos candidatos, um composto chamado MCB-613 destacou-se de forma consistente. Células resistentes que ignoravam os inibidores de EGFR mostraram-se incomumente vulneráveis ao MCB-613, tanto em placas de cultura quanto em tumores de camundongos.
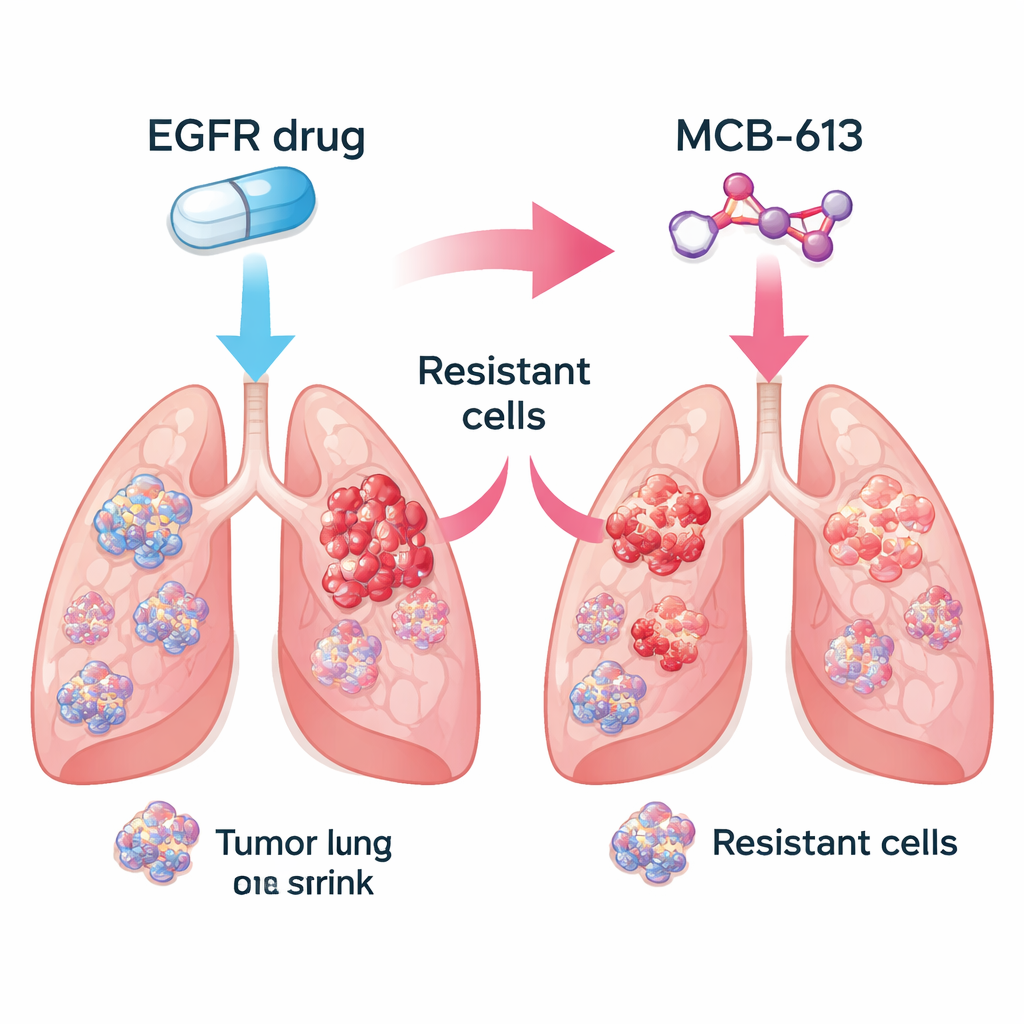
Encurralando populações tumorais mistas Figure 1.
Tumores reais são misturas de células: algumas permanecem sensíveis ao medicamento original, enquanto outras adquirem resistência por meio de diferentes artifícios genéticos. A equipe perguntou se combinar um inibidor de EGFR com MCB-613 poderia eliminar essa diversidade. Em um experimento controlado, eles misturaram principalmente células sensíveis com uma pequena fração de vários tipos resistentes, imitando o tumor de um paciente. Tratar essa população mista com o inibidor de EGFR ou com MCB-613 isoladamente permitiu que algumas células sobrevivessem e crescessem. Mas quando ambos agentes foram usados em conjunto, toda a população colapsou. Isso sugere que parear uma terapia alvo padrão com um fármaco de “sensibilidade colateral” cuidadosamente escolhido pode empurrar tumores para um beco evolutivo sem saída.
Uma ponte molecular que quebra um guardião
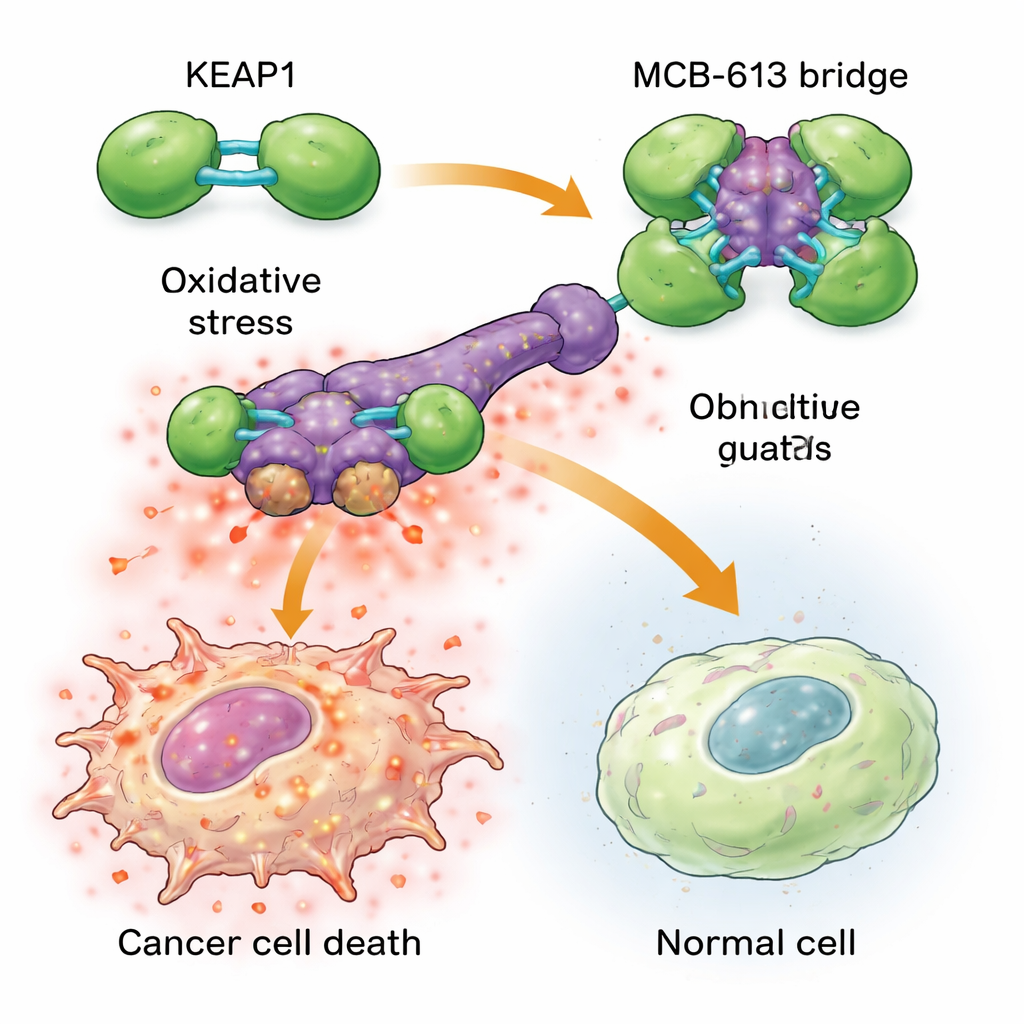
Para entender por que o MCB-613 atinge as células resistentes com tanta força, os cientistas examinaram a quais proteínas ele se liga. Usando sondas químicas e uma triagem dirigida por CRISPR, identificaram uma proteína chamada KEAP1 como essencial para o efeito do MCB-613. KEAP1 normalmente atua como um guardião celular, detectando estresse e ajudando a regular respostas protetoras. A equipe descobriu que o MCB-613 se liga ao KEAP1 de uma maneira incomum: comporta-se como uma ponte molecular rígida que conecta unidades de KEAP1 entre si em agregados exagerados e anormais. Esse processo não depende dos habituais locais reativos contendo enxofre no KEAP1, mas sim de uma lisina específica na região de dimerização. Quando essa lisina foi mutada, o MCB-613 não conseguiu mais agrupar o KEAP1, e as células resistentes deixaram de ser hipersensíveis ao composto.
Transformando estresse útil em sobrecarga letal Figure 2.
A agregação do KEAP1 desencadeia uma cadeia perigosa de eventos dentro das células cancerígenas resistentes a drogas. Essas células já vivem sob um estresse basal mais alto, com níveis elevados de espécies reativas de oxigênio (subprodutos químicos danosos) e atividade aumentada em uma rede protetora conhecida como resposta integrada ao estresse. Quando o MCB-613 é adicionado, a disrupção do KEAP1 empurra esse estado estressado além do limite: o oxigênio reativo se acumula ainda mais, e reguladores-chave do estresse chamados ATF4 e CHOP ligam programas potentes de morte. Bloquear esses reguladores do estresse, ou quimicamente neutralizar espécies reativas de oxigênio, protegeu em grande parte as células contra o MCB-613. Curiosamente, o parceiro clássico do KEAP1, NRF2, frequentemente considerado o principal condutor das defesas antioxidantes, não foi responsável pela morte; na verdade, remover NRF2 tornou as células ainda mais sensíveis, ressaltando que o MCB-613 explora uma via não canônica e diferente.
O que isso pode significar para tratamentos futuros
Por ora, o próprio MCB-613 é um composto-ferramenta com desvantagens químicas que o tornam inadequado como medicamento. Mas ele revela um conceito poderoso: à medida que cânceres de pulmão evoluem resistência a inibidores de EGFR, podem ficar travados em um estado estressado que pode ser seletivamente atacado por compostos que forçam o KEAP1 a formar montagens disfuncionais. Em princípio, versões refinadas dessas “pontes moleculares” poderiam ser desenvolvidas para serem mais seguras e precisas, dando aos oncologistas uma forma de conduzir tumores a uma “escolha impossível” entre sensibilidade à terapia alvo original e sensibilidade ao agente indutor de estresse subsequente. Essa estratégia de aprisionamento evolutivo poderia, eventualmente, ajudar a atrasar ou superar a resistência em cânceres de pulmão com mutação em EGFR e possivelmente em outros cânceres de difícil tratamento também.
Citação: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1
Palavras-chave: câncer de pulmão com mutação em EGFR, resistência a fármacos, sensibilidade colateral, KEAP1, estresse oxidativo